Object description
Here we review the structure, intramolecular bonds and ligand interactions of E. coli lactaldehyde reductase (FucO) M185C mutant (PDB ID 5BR4) using the structure visualisation published in Protein Data Bank (Cahn et al., 2016)[4].
Lactaldehyde reductase is an enzyme, belonging to the family of oxidoreductases (EC number 1.1.1.77). Another name is propanediol:NAD+ oxidoreductase. It calyzes the reaction: (R)[or (S)]-propane-1,2-diol + NAD+ ↔ (R)[or (S)]-lactaldehyde + NADH + H+, during which 2 electrons are transfered from propane-1,2-diol to NAD+, the former converts to lactaldehyde losing 2 H+ and the latter converts to NADH gaining 1 H+. The substrates are (R)-propane-1,2-diol or (S)-propane-1,2-diol (the reaction is non-stereoselective) and NAD+. It can participate in propanoate[5], glyoxylate and dicarboxylate metabolism[6].
Discription of native protein structure (PDB ID 1RRM):
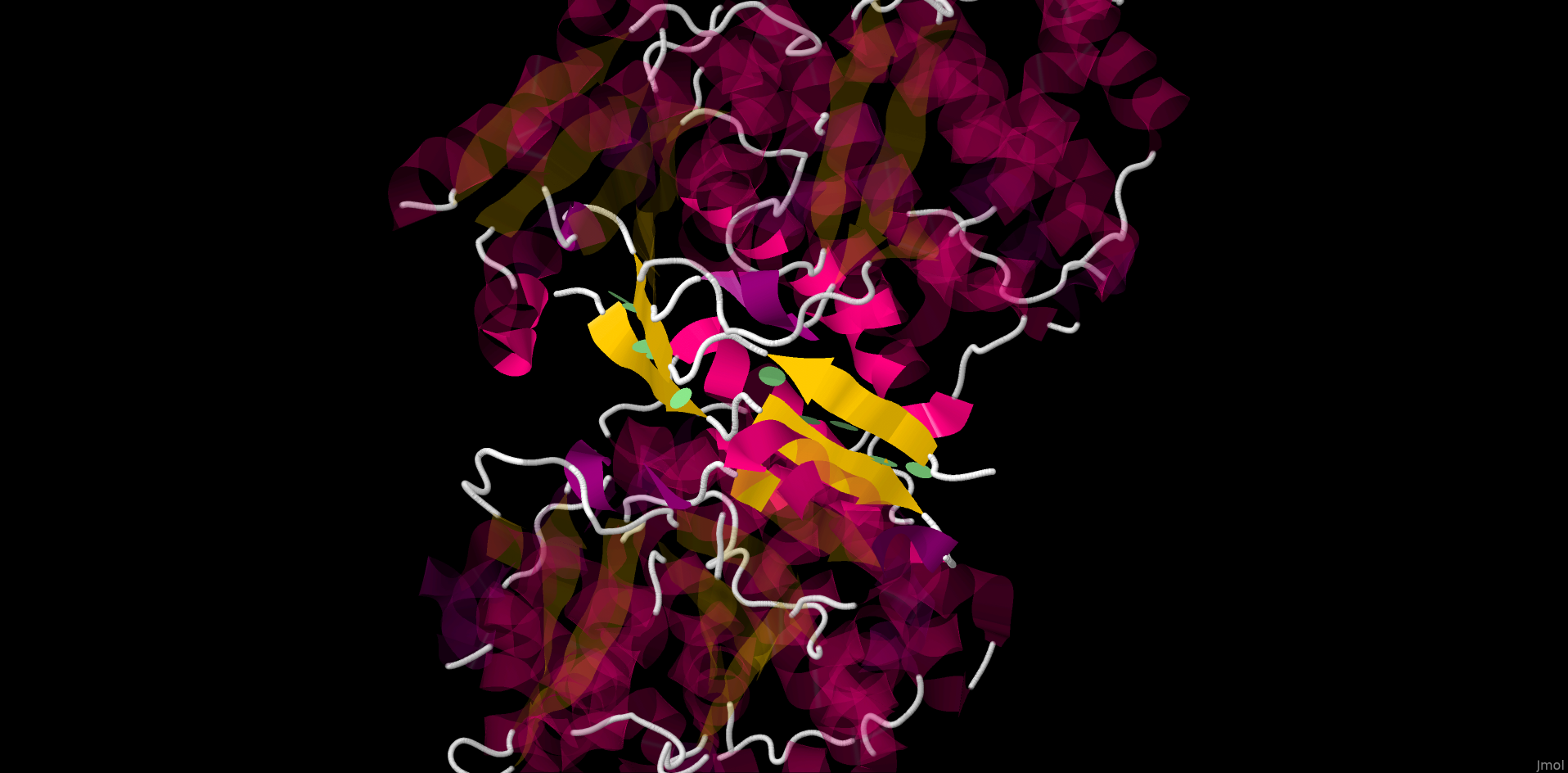
This is a globular protein found in bacterial cytoplasm. The molecule is a homodimer, consisting of 2 chains (A, B), held together by 10 hydrogen bonds between 2 antiparallel strands from different chains, forming 2 β-sheets (each chain has 2 β-strands linked by short unstructured motif). Each chain has 1 active site homing Zn2+ ligand. Ajacent to it lie propanediol/lactaldehyde- and NAD+/NADH-binding sites. The latter includes DHQS-like fold (named after 3-dehydroquinate synthase) - one of the structures abundant in NAD(P)H-binding enzymes, consisting of alternating β-strands and α-helixes.
Description of the current protein structure and properties (PDB IB 5BR4):
This mutant form is interesting for being one of the rare cases, when a mutation improves the enzyme catalytic activity in native reaction without changing the enzyme selectivity, general structure or other physico-chemical properties. Usually mutations, altering enzymes catalytic activity, are associated with active site structure modification, resulting in unpredictably changing substrates affinities and even the main catalysing reaction; such characteristics as optimal pH and temperature often may also be affected. In vitro and in vivo tests showed the mutation hadn't changed considerably any protein property, besides increasing its activity in its native reaction: thermostability didn't change and bacteria with such mutation became even more durable to toxic aldehydes. The mutation replaces methionine with cysteine in 185 position of both chains, the corresponding residue is situated distal to the active site in DHQS-like motif of NAD-binding site. Cysteine is more polar than methionine and, though its side chain is almost aligned with β- and γ-C atoms of methionine and there are no major changes in NAD-binding site structure, the mutation slightly shifts adenine of NADH by 0,3Å toward active site. And the adenine position change results in a more significant shift of 1.1Å at the N1 atom of the nicotinamide at the other end of the cofactor. As nicotinamid N-1 directly takes part in the reaction by accepting electrons, it is assumed that its shift toward active site plays the main role in enhancing enzyme catalytic activity[4].
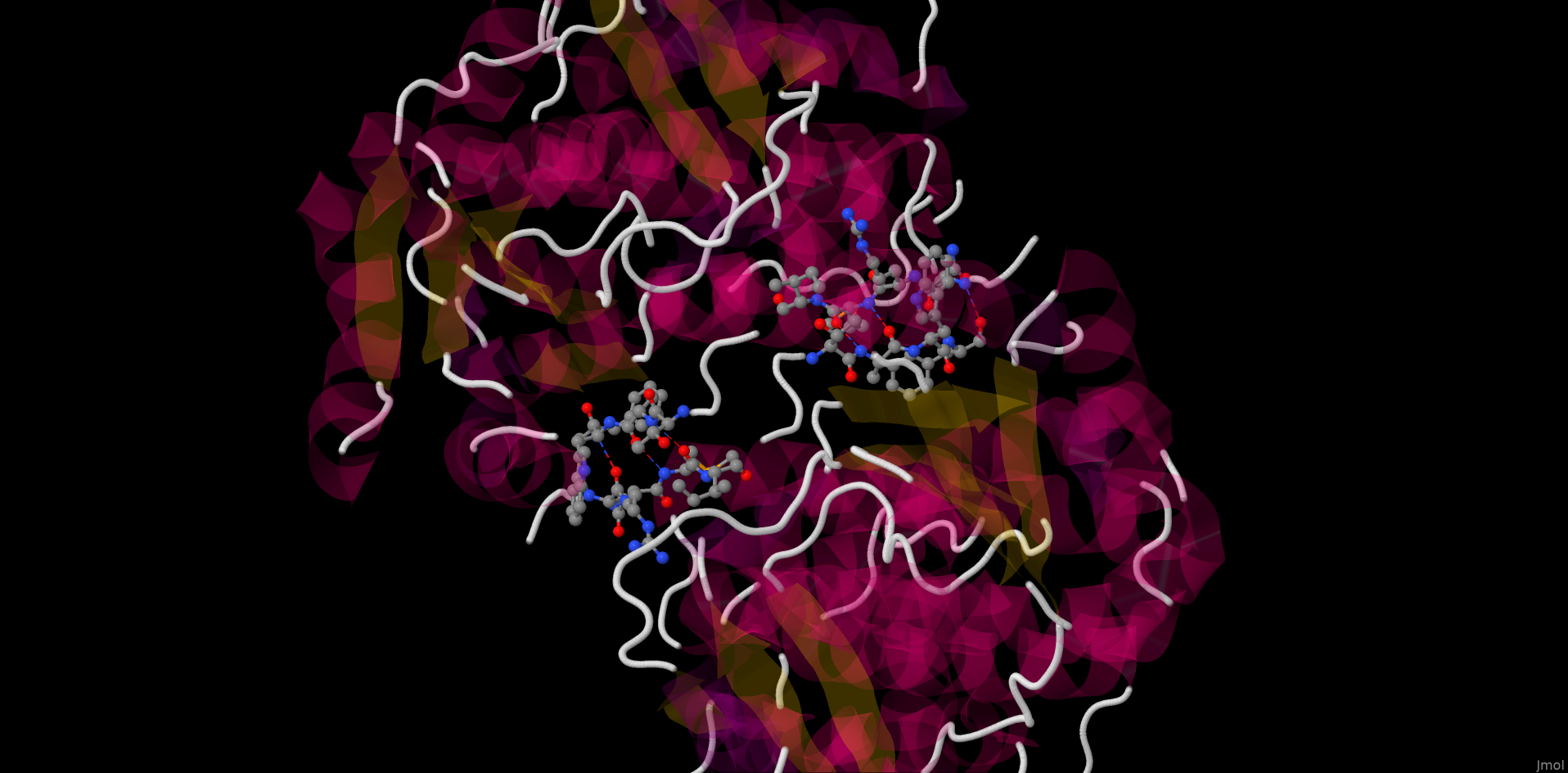
Chosen preparation methods of protein crystal dramatically influenced the protein quaternary structure and its ligand composition. The use of NH4Cl as precipitant resulted in lots of chloride ions embedded into the protein, which obviously have no catalitic effect, as being absent in the native form, and do not cause any significant structure altering. Glycerol was used for protein soaking. Being structurally similar to propanediol, it integrated into propanediol/lactaldehyde-binding site. Unlike chloride ions, glycerol changed the protein structure by intercalating between the 2 chain globules, rotating them, and linking by forming hydrogen bond-based glycerol bridge just at the opposite to native structure β-strands globules poles. So in corresponding crystal the protein ligands includes 2 NADH molecules, 3 glycerol molecules (2 near the active sites and 1 linking the chains) and multiple chloride ions, the protein chains are held together by a glycerol bridge[4].
Brief description of ligands
|
 |
||||||||||
|
 |
||||||||||
|
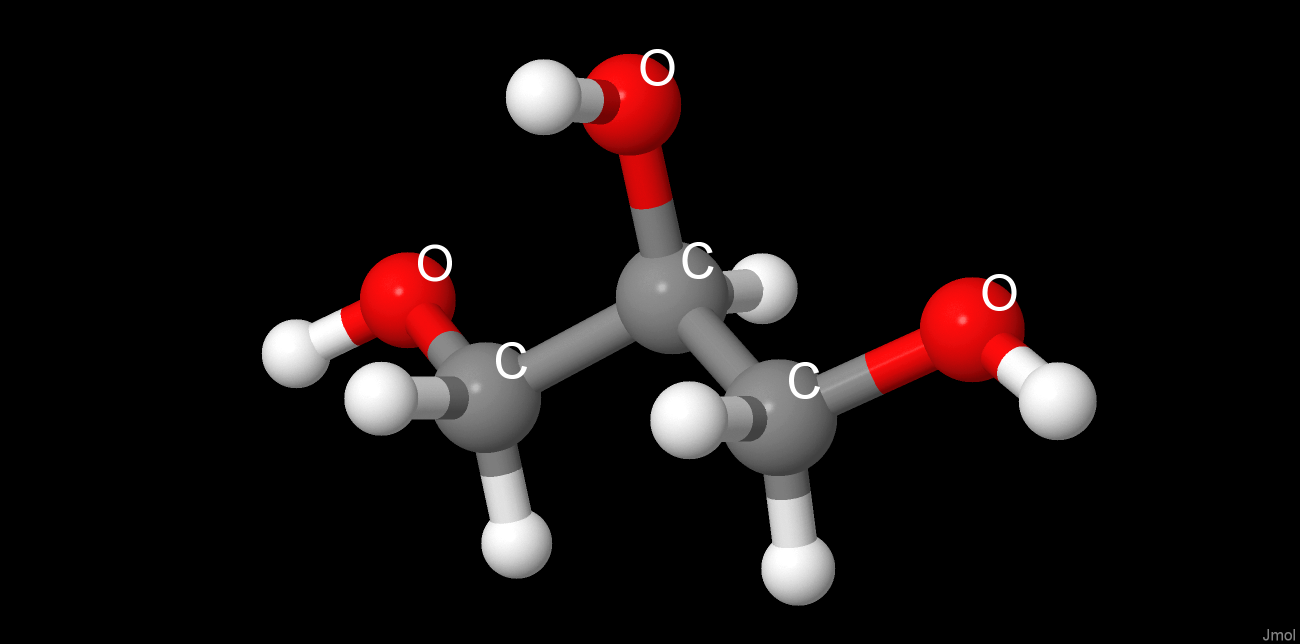 |
||||||||||
|
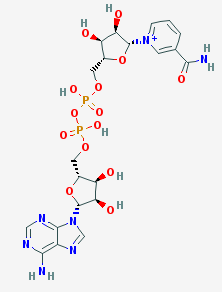
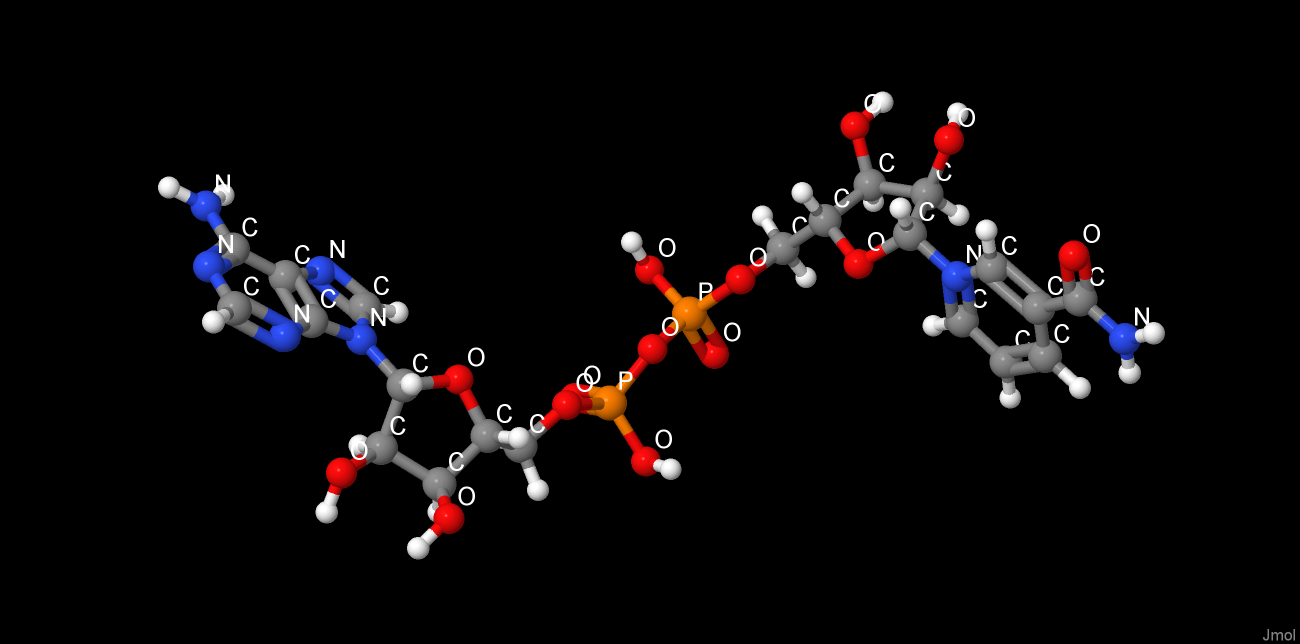 |
Interaction description
Protein-protein interactions
Using JMol we managed to reveal only hydrogen bonds via the glycerol bridge between the 2 subunits. However, in water solution and in vivo the protein quarternary structure is defined by different interactions. To determine these interactions and protein native structure we reviewed the model of lactaldehyde reductase crystalized from water solution (PDB ID 1RRM). Applying J(S)Mol comands "contact" and "hbond calculate" we found 10 interchain hydrogen bonds shown in the picture below.
Protein-ligand interactions
Coordinate bonds with zinc cation
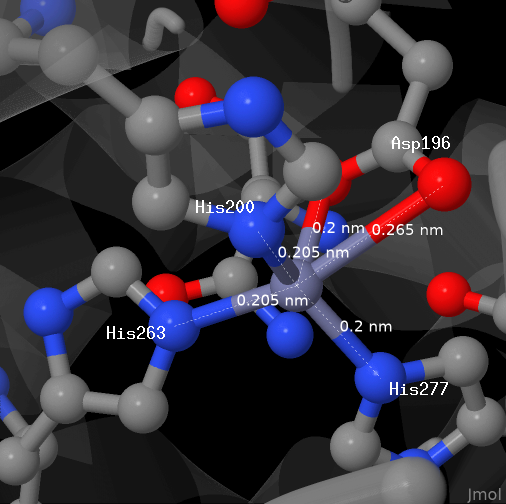
The picture above dipicts zinc cation from the studied protein (chain A). The cation is bond with 4 aminoacid residues by coordinate bonds.
Each of the aspartate residue anion oxygens forms a bond with the cation.
All the bonds are shown in PDB and displayed here as default.
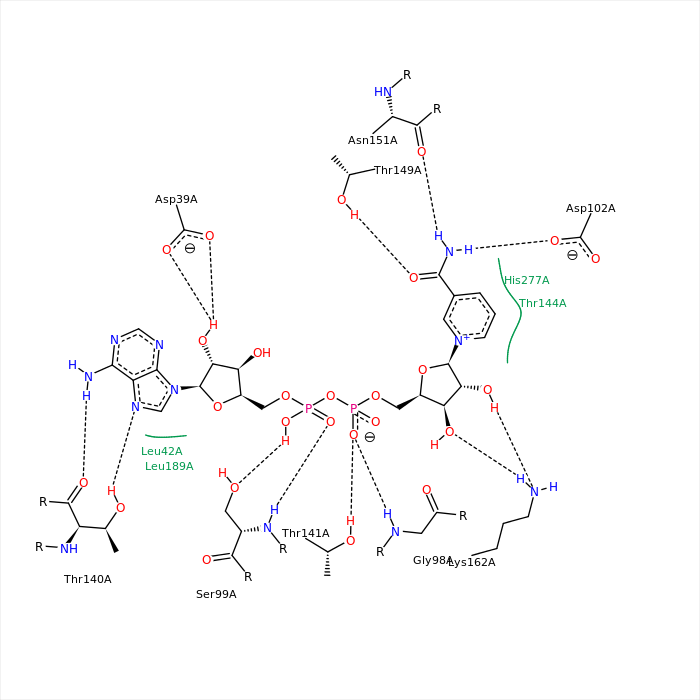
The picture above depicts NAD from the studied protein (chain A). The picture is imported from rcsb.org. Hydrogen bonds were found with the help of J(S)Mol command "hbonds calculate" with RasMol disabled. The bonds lengths range between 2,75-3,24Å.
Hydrophobic nucleus
For hydrophobic nuclei detection we used the CluD programm. Nuclei with more than 5 atoms were chosen. As the protein is a homodimer, the nuclei are situated simmetrically, 3 in each chain.
We chose leucine residue 284:A from hydrophobic nucleus №1 as MyResidue. As it can be seen in applet, atoms, situated in 6Å from MyResidue, almost entirely cover it. To determine the average distance between hydrophobic nucleus atoms, which are not covalent-bound, we made 20 measurements, used to build the table. In calculations we used the following Van der Waals radii values: carbon - 1,85Å, nitrogen - 1,54Å, oxygen - 1,4Å, sulfur - 1,85Å.[1]
| # | Distance between the atoms centres, Å |
Atom #1 coordinates | Atom #2 coordinates | Distance between the atoms Van der Waals surfaces, Å |
| 1 | 3,53 | [MET]265:A.CE | [TYR]379:A.CG | -0,17 |
| 2 | 3,88 | [TYR]379:A.CD1 | [LEU]340:A.CD1 | 0,18 |
| 3 | 3,66 | [LEU]340:A.CD2 | [ILE]283:A.CG2 | -0,04 |
| 4 | 4,09 | [VAL]288:A.CG2 | [MET]265:A.SD | 0,39 |
| 5 | 3,85 | [LEU]269:A.CD1 | [LEU]340:A.CD2 | 0,15 |
| 6 | 3,80 | [HIS]287:A.CD2 | [ALA]382:A.CB | 0,10 |
| 7 | 3,38 | [ASN]281:A.O | [LEU]285:A.CB | 0,13 |
| 8 | 3,55 | [ALA]382:A.CB | [LEU]378:A.O | 0,30 |
| 9 | 3,38 | [ILE]283:A.O | [HIS]287:A.CD2 | 0,13 |
| 10 | 3,45 | [ALA]282:A.O | [PRO]286:A.CD | 0,20 |
| 11 | 2,43 | [ASN]281:A.N | [ALA]280:A.CA | -0,96 |
| 12 | 3,85 | [LEU]269:A.CD1 | [LEU]340:A.CD2 | 0,15 |
| 13 | 5,16 | [ALA]382:A.CB | [LEU]284:A.CA | 1,46 |
| 14 | 4,18 | [LEU]284:A.CD1 | [LEU]269:A.CD1 | 0,48 |
| 15 | 4,55 | [LEU]284:A.CG | [LEU]340:A.CD2 | 0,85 |
| 16 | 3,94 | [LEU]284:A.CD1 | [ALA]266:A.CA | 0,24 |
| 17 | 4,25 | [LEU]284:A.CD2 | [ALA]382:A.CB | 0,55 |
| 18 | 4,44 | [LEU]284:A.CD2 | [MET]265:A.SD | 0,74 |
| 19 | 3,82 | [LEU]284:A.CD2 | [MET]265:A.CE | 0,12 |
| 20 | 4,54 | [LEU]284:A.CB | [VAL]288:A.CG2 | 0,84 |
Average distance between atoms Van der Waals surfaces in the hydrophobic nucleus is 0,3Å, that is surely insufficient to embed there an oxygen atom with radius 1,4Å without electron clouds overlapping. Average internuclei distance is 3.89Å, so it allows the oxygen atom to be placed there with its electron shell being overlapped with those of other hydrophobic nucleus atoms.
Authors contribution
Pushkarev Sergey was responsible for searching and describtion of the hydrophobic nuclei, ligands searching, corresponding scripts tuning and writing the review script. Bushmakin Ilya was responsible for description of coordinate and hydrogen bonds between the protein and ligands (zinc and NAD), description of intrachain amino-aromatic interactions, the web-site arrangement. Puhov Stepan was responsible for literature studying, description of the protein, its structure and function, alternative protein structures searching, the web-site translation.
References
- Дж, Эмсли. Элементы. М.: Мир, 1993.
- Burley, S. K., and G. A. Petsko. "Amino‐aromatic interactions in proteins." FEBS letters 203.2 (1986): 139-143.
- Dougherty, Dennis A. "Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp." Science 271.5246 (1996): 163-168.
- Cahn, J.K.B. et al. “Mutations in Adenine-Binding Pockets Enhance Catalytic Properties of NAD(P)H-Dependent Enzymes.” Protein Engineering, Design and Selection 29.1 (2016): 31–38. PMC. Web. 7 Mar. 2018.
- "KEGG PATHWAY: map00640",KEGG: Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg.
- "KEGG PATHWAY: map00630",KEGG: Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg.