Анализ качества
| Команда | Описание |
|---|---|
fastqc chr12.fastq |
Выдает график оценки качества нуклеотидов и некоторую другую информацию о последовательности. Выполнялась дважды. |
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr12.fastq trimmed12.fastq SLIDINGWINDOW:10:28 MINLEN:50 |
Обрезает концы плохого качества (по 10 со средним качеством ниже 28, оставляя только прочтения >50bp). |
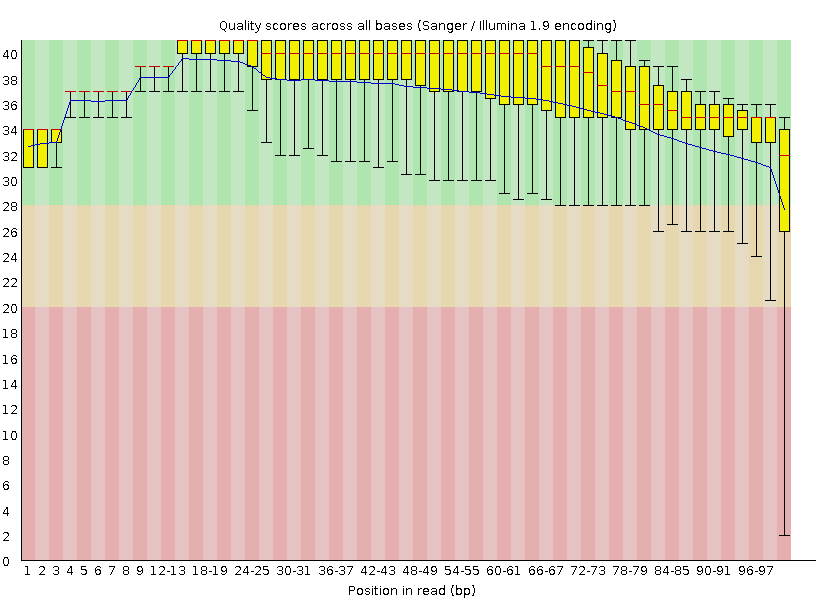
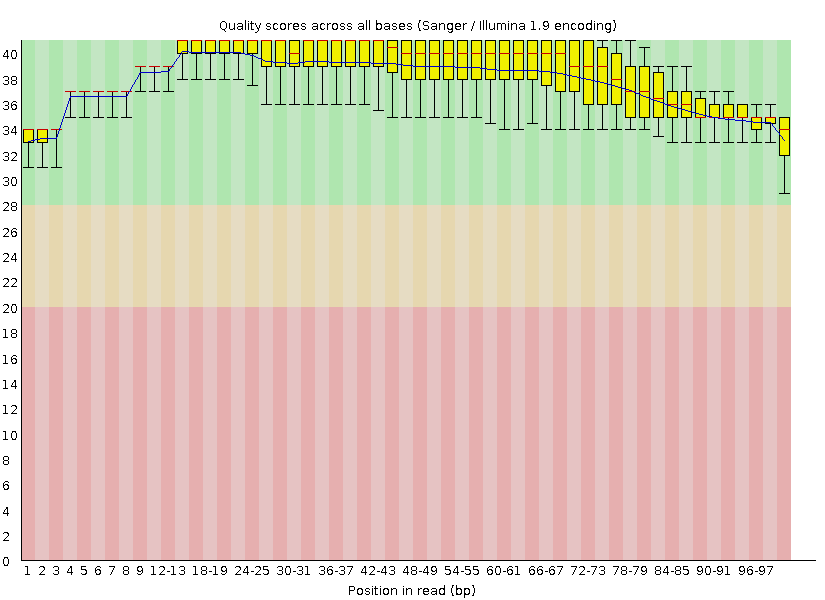
Количество прочтений уменьшилось после чистки с 7157 до 6427. Отсеялись непопавшие под критерии риды (критерии в таблице команд).
Картирование чтений
| Команда | Описание |
|---|---|
hisat2-build chr12.fasta chr12 |
Индексирует последовательность. |
/home/students/y06/anastaisha_w/hisat2-2.0.5/hisat2 \ chr12 trimmed12.fastq -S alg_chr12.sam --no-spliced-alignment --no-softclip |
Строит выравнивание рида и референса. |
samtools view -b -o alg_chr12.bam alg_chr12.sam |
Перевод выравнивания в бинарный формат. |
samtools sort alg_chr12.bam sorted_chr12 |
Сортирует выравнивания по координатам начала. |
samtools index sorted_chr12.bam |
Индексирует п |
samtools depth -r chr12:66260584-66260734 sorted_chr12.bam > depth_exon.tsv |
Выводит таблицу покрытий для каждого нуклеотида в указанном диапазоне. |
Всего очищенный файл содержит 6427 рида, все непарные. 15 из них не выравнены, остальные ровно один раз.
Для анализа экзонов взят экзон гена AK128707 (неизвестный белок) chr12:66'260'584..66'260'734. Среднее покрытие 139.8, максимум покрытия примерно в середине, как и ожидалось.
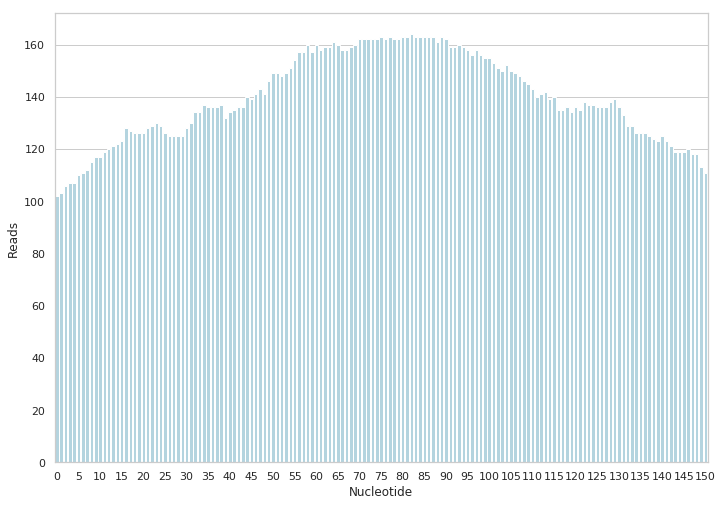
Полиморфизмы
| Команда | Описание |
|---|---|
samtools mpileup -uf chr12.fasta -o poly.bcf sorted_chr12.bam |
Формирует бинарный файл с полиморфизмами. . |
bcftools call -cv poly.bcf -o poly.vcf |
Переводит файл с полиморфизмами в почти человекочитаемый формат. |
convert2annovar.pl -format vcf4 poly.vcf > poly.avinput |
Переводит файл с полиморфизмами vcf в формат annovar. |
annotate_variation.pl -filter -out snp138_filtered -build hg19 -dbtype \ snp138 poly.avinput /nfs/srv/databases/annovar/humandb.old/ |
Разделяет полиморфизмы по наличию в dbsnp. |
annotate_variation.pl -out refGene_filtered -build hg19 poly.avinput \ /nfs/srv/databases/annovar/humandb.old/ |
Аннотация refGene. Выделяет положение полиморфизма и вносимые изменения, если она в экзоне. |
annotate_variation.pl -filter -out 1000g -build hg19 -dbtype 1000g2014oct_all \ poly.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация 1000 genomes. Показывает частоты аллелей. |
annotate_variation.pl -regionanno -out gwas -build hg19 -dbtype gwasCatalog \ poly.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация GWAS. Выделяет ассоциированные признаки |
annotate_variation.pl -filter -out clinvar -build hg19 -dbtype clinvar_20140211 \ poly.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация ClinVar. Должен выделять ассоциации с болезнями. |
| Координаты | Тип | Изменения | Глубина | Качество прочтений |
|---|---|---|---|---|
| 9'822'387 | Замена (трансверсия) | C->G | 28 | 174.009 |
| 66'260'925 | Замена (трансверсия) | C->G | 4 | 30.0167 |
| 107'367'224 | Замена (транзиция) | T->C | 74 | 221.999 |
Обнаружены только замены (всего 33, 7 не имеют rs, все приведенные уже в базе) |
||||
Почти у всех полиморфизмов хорошее покрытие. Интересно, что именно полиморфизмы с плохим покрытием отсутствуют в dbSNP и 1000genomes. Возможно, это артефакты секвенирования.
RefSeq в annovar делит SNP по положению (в скобках - сколько в данной последовательности): exonic(3), splicing(0), intronic(23), ncRNA(1), UTR5(0), UTR3(6), upstream, downstream, intergenic. Названия говорящие. Все SNP попали в 4 гена: CLEC2D(natural killer C-type receptor member, в этом гене все экзонные SNP), RPSAP52(Ribosomal Protein SA Pseudogene 52), HMGA2 (High modility group protein, DNA binding, нокаут ассоциирован с ожирением у мышей), TMEM263.
ClinVar не содержит информации по полиморфизмам. GWAS содержит информацию о 4 SNP: CLEC2D - диабет первого типа, HMGA2 - рост и структура мозга, TMEM263 - минерализация костей.