Чистка и анализ качества чтений
Для удаления адаптеров создан отдельный файл со всеми адаптерами с помощью seqret. Для оценки качества очистки составлялся отчет fastqc до и после очистки.
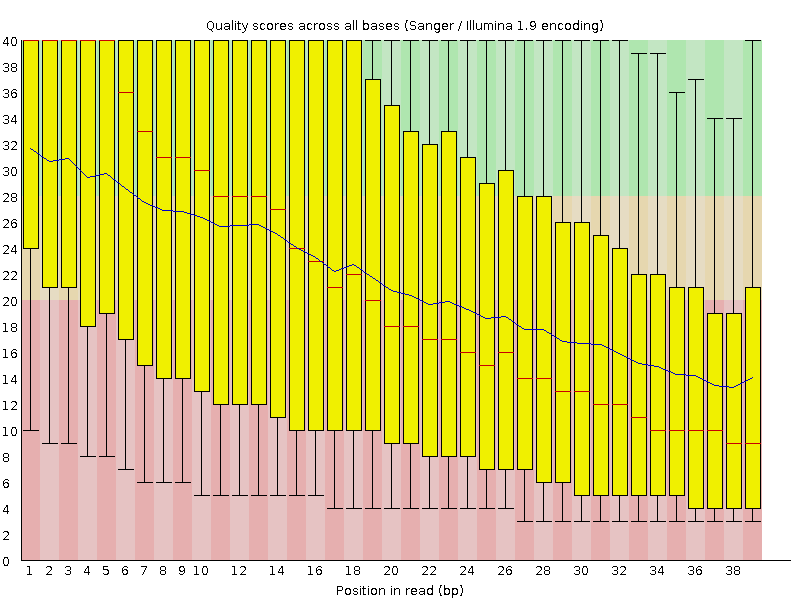
Очистка проводилась командой (удаление адаптеров, удаление концов с качеством ниже 20, минимальная длина 30 bp). Число чтений уменьшилось с 10'543'839 до 8'580'909, удалено 1'962'930. Файл уменьшился с 1.2 Гб до 919 Мб.
java -jar \ /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE \ -phred33 SRR4240358.fastq cropped.fastq ILLUMINACLIP:adapters.fasta:2:7:7 \ TRAILING:23 MINLEN:30
Сборка
Сборка проводилась командами: Final graph has 698 nodes and n50 of 12287, max 38496, total 661317, using 0/8580909 reads
velveth asmbl 29 -fastq cropped.fastq velvetg asmbl
| Длина k-меров | ||
|---|---|---|
| 25 | 29 | |
| Контиги (Nodes) | 8227 | 698 |
| N50 | 1576 | 12'287 |
| Максимальная длина | 12'273 | 38'496 |
Анализ 3 контигов
Анализировались 3 контига с длинами 38496, 30747, 27984.
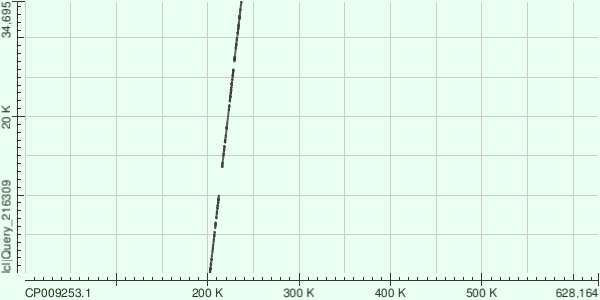
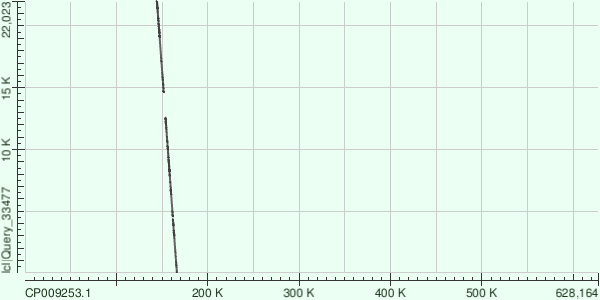
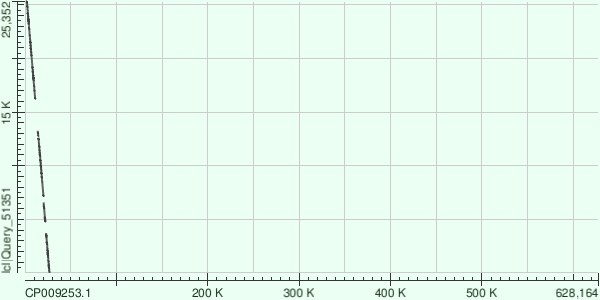
Выравнивания оставляют желать лучшего. Странно, что простой поиск megablast по nr банку дает выравнивания с 99% идентичности с геномом других штаммов. Например, Node 9 выравнивается с Buchnera aphidicola str. 5A 99%.
Плохое выравнивание может быть объяснено плохим качеством ридов или сильной внутривидовой вариативностью бактерии.