Для поиска всех последовательностей, полученных методом электронной микроскопии, в Advanced search в качестве Experimental method (сначала Add search criteria) был выбран Electron microscopy. Найдено было всего 929 структур. С помощью Download results получен fasta-файл, содержащий последовательности всех белков.
Для 5E4R.A (domain-duplicated synthetic class II ketol-acid reductoisomerase) был найден дальний структурный гомолог 2IZZ.A (pyrroline-5-carboxylate reductase 1). С помощью сервиса RCSB PDB Protein Comparison Tool алгоритмом jFatCat были построены гибкое (RMSD: 2.64; 10,5%) и жесткое (RMSD: 3.40; 8.6%) структурные выравнивания. Результаты выравнивания приведены на рис.1.
Рисунок 1. Пространственное наложение структур

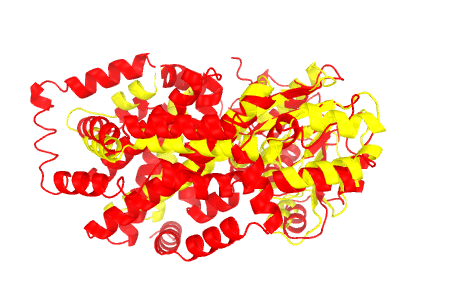
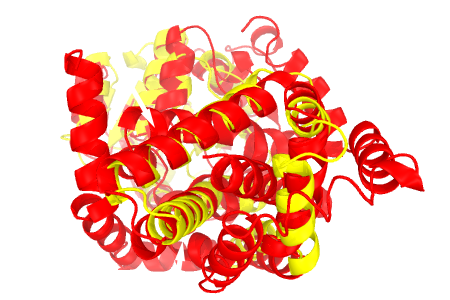
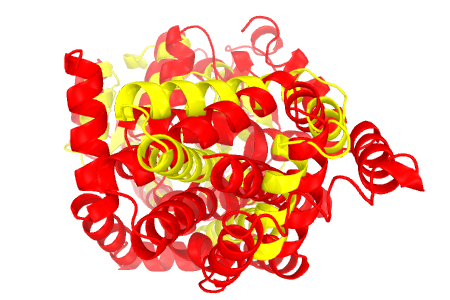
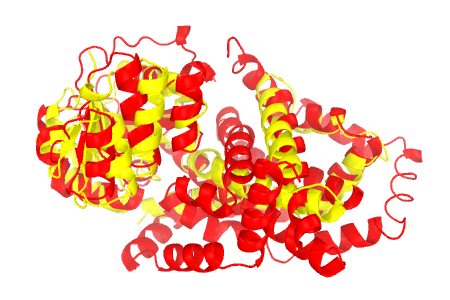
2IZZ.A показана желтым, 5E4R.A - красным. Молекулы изображены с разных ракурсов (A/B/C) для демонстрации различий в выравнивании. Слева расположены изображения гибких выравниваний, справа - жестких. Видно, что в случае гибкого выравнивания большее количество спиралей накладывается друг на друга.
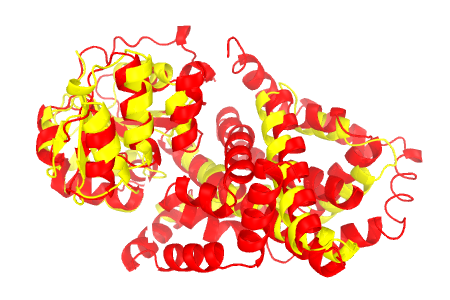
Другой пример: 1I2A(RIBOSOMAL PROTEIN L1P) и 1AD2(RIBOSOMAL PROTEIN L1). Гибкое (RMSD: 2.19; 26.3%) и жесткое (RMSD: 3.81; 15.8%) выравнивания показаны на рис.2. Здесь разница и в RMSD, и в доле выравненных остатков более значительно при разных способах структурного выравнивания.
Рисунок 1. Пространственное наложение структур
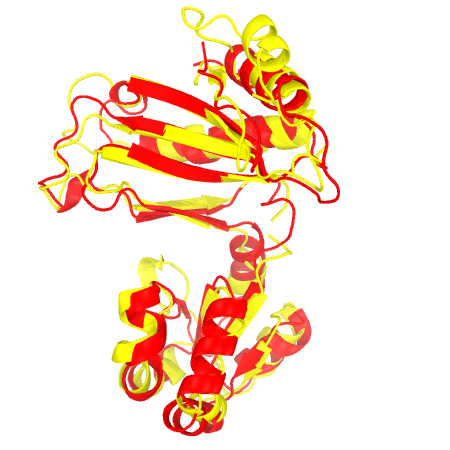
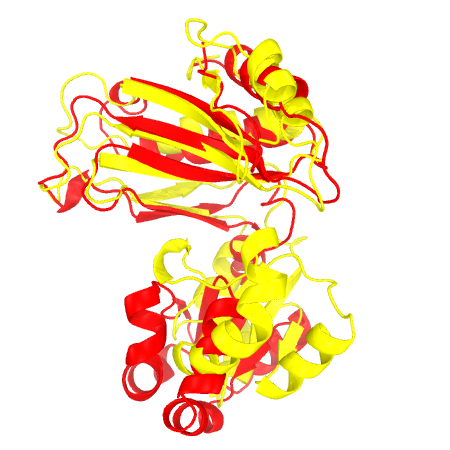
1I2A.A показана красным, 1AD2.A - желтым. Слева - гибкое выравнивание, справа - жесткое.
Для демонстрации более четкой разницы в работе алгоритмов можно использовать структуры одного и того же белка в разных конформационных состояниях. Тогда гибкое выравнивание дает значительно лучший результат.
PS: все изображения получены в PyMol.