В первую очередь, соберем файл с последовательностями адаптеров, чтобы удалить их из последовательности файла.
Далее проведём триммирование чтений:
Прежде рассмотрим выдачу файла: Input Reads: 7400155 Surviving: 7269852 (98.24%) Dropped: 130303 (1.76%). Таким образом 1.76% чтений оказались с частями адаптеров. Следующим этапом удалим нуклеотиды с плохим качеством (менее 20), и чтения с неудовлетворяющей нас длиной (менее 32).
Выдача файла:Input Reads: 7269852 Surviving: 6974267 (95.93%) Dropped: 295585 (4.07%). В результате удалено еще 295585 чтений (4.07% от оставшегося числа).
Размеры файлов, после использованных программ: SRR4240379.fastq.gz (174Mb); SRR4240379_clean.fastq.gz (172 Mb); clea_trim.fasta.gz (162 Mb).
Подготавливаем необходимые k-меры:
В конце на выдаче получаем такие данные:Final graph has 440 nodes and n50 of 25646, max 49912, total 664650, using 0/6974267 reads. Для себя отмечаем, что N50 = 25646. Чтобы определить какие значения выбиваются из общего порядка, найдем медиану. Определив количество контигов, нахожу медиану и ее значения (>NODE 194 length 45 cov 16.555555)
Наиболее длинные
Покритие превышающее более чем в 5 раз медиану
Покритие более чем в 5 раз меньше медианы
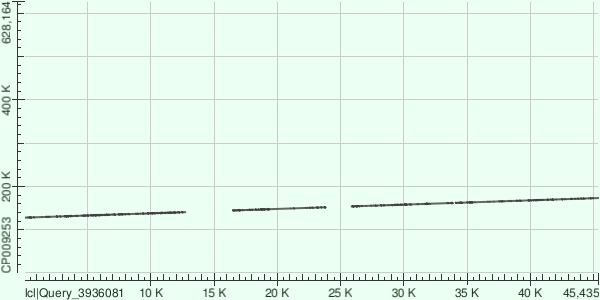
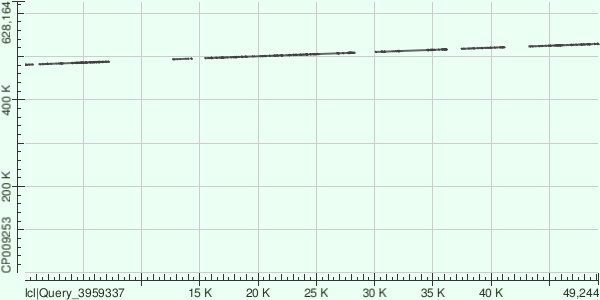
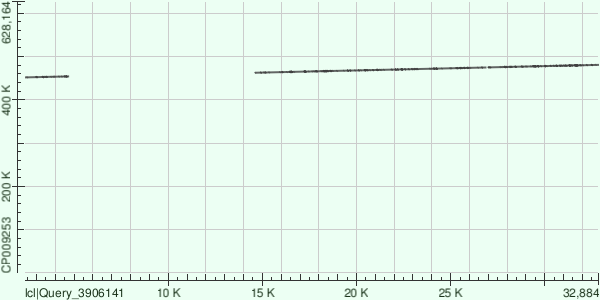
Для построения Dot Plot выделили последовательности контигов, выравнивание воспроизводилось относительно хромосомфы Buchnera aphidicola (GenBank/EMBL AC — CP009253).