Практикум 13. Алгоритмы и программы множественного выравнивания
Сравнение выравниваний одних и тех же последовательностей разными программами
Для последующего анализа сравнений выравниваний были выбрали последовательности с доменом Glycoside hydrolase 97, который был выбран в практикуме 11: Probable retaining alpha-galactosidase (D7CFN7), Glucan 1,4-alpha-glucosidase SusB (G8JZS4), Retaining alpha-galactosidase (Q8A6L0), Glycoside hydrolase 97 family protein (A0A031I7D5), Alpha-glucosidase (A0A066WK53).
Было проведено множественное выравнивания этих пяти последовательностей программами MAFFT, MUSCLE, TCOFFE и CLUSTALW. Затем с помощью программы, которая была сделана моими однокурсниками, был осуществлен анализ сравнения выравниваний. В качестве программы референсного выравнивания была выбрана программа MAFFT. Оставшиеся выравнивания сравнивались именно с ней.
Проект Jalview с выравниваниями (первые 5 строк — MUSCLE, вторые 5 строк — MAFFT, третьи 5 строк — TCOFFEE, последние — ClustalW).
1. Сравнение MAFFT и MUSCLE. Длина выравнивания MAFFT — 865, MUSCLE — 831. Процент совпадающих колонок у MAFFT был равен 55,84%, у MUSCLE — 58,12%. Результат представлен в таблице 1
| Block | MAFFT | MUSCLE |
|---|---|---|
| 1 | 42-48 | 33-39 |
| 2 | 70-91 | 77-98 |
| 3 | 117-120 | 123-126 |
| 4 | 131-158 | 137-164 |
| 5 | 170-180 | 176-186 |
| 6 | 192-211 | 198-217 |
| 7 | 257-261 | 252-256 |
| 8 | 270-304 | 264-298 |
| 9 | 307-309 | 301-303 |
| 10 | 332-340 | 324-332 |
| 11 | 351-405 | 338-392 |
| 12 | 411-419 | 398-406 |
| 13 | 434-443 | 421-430 |
| 14 | 451-461 | 438-448 |
| 15 | 485-488 | 466-469 |
| 16 | 499-515 | 482-498 |
| 17 | 551-569 | 534-552 |
| 18 | 573-578 | 556-561 |
| 19 | 582-631 | 565-614 |
| 20 | 641-670 | 621-650 |
| 21 | 694-755 | 670-731 |
| 22 | 763-801 | 739-777 |
| 23 | 827-842 | 798-813 |
Как видно из таблицы 1, общее количество совпадающих блоков равно 23. Сдвиг при выравнивании колонок произошел сразу на первой позиции, поэтому все последующие блоки оказались не равными между собой. Список одинаково выровненных колонок вне блока (первое число — колонка в выравнивании MAFFT, второе — MUSCLE): 164-170, 263-258, 426-413, 464-451, 691-667, 813-784.
2. Сравнение MAFFT и CLUSTALW. Длина выравнивания MAFFT — 865, CLUSTALW — 816. Процент совпадающих колонок для выравнивания MAFFT — 51,56%, CLUSTALW — 54,66%. Результат представлен в таблице 2.
| Block | MAFFT | CLUSTALW |
|---|---|---|
| 1 | 1-4 | 1-4 |
| 2 | 42-48 | 44-50 |
| 3 | 69-91 | 71-93 |
| 4 | 131-180 | 132-181 |
| 5 | 269-291 | 264-286 |
| 6 | 294-312 | 289-307 |
| 7 | 339-350 | |
| 8 | 367-385 | 355-373 |
| 9 | 389-401 | 377-389 |
| 10 | 451-461 | 436-446 |
| 11 | 551-560 | 520-529 |
| 12 | 564-569 | 533-538 |
| 13 | 573-631 | 542-600 |
| 14 | 645-670 | 611-636 |
| 15 | 694-723 | 656-685 |
| 16 | 726-778 | 688-740 |
| 17 | 788-800 | 750-762 |
| 18 | 827-844 | 784-801 |
В представленом сравнении выравниваний оказалось меньше всего блоков по количеству. Однако процент выровненных колонок не уступает Muscle и T-Coffee. Список одинаково выровненных колонок вне блока (первое число — колонка в выравнивании MAFFT, второе — ClustalW): 98-100, 426-414, 433-421, 812-769, 848-805.
3. Сравнение MAFFT и T-COFFEE. Длина выравнивания MAFFT — 865, T-COFFE — 837. Процент совпадающих колонок у программы MAFFT был равен 54,80%, в то время как у T-COFFE — 56,63%. результаты представлены в таблице 3.
| Block | MAFFT | T-COFFEE |
|---|---|---|
| 1 | 39-41 | 39-41 |
| 2 | 43-48 | 43-48 |
| 3 | 50-52 | 50-52 |
| 4 | 69-91 | 69-91 |
| 5 | 131-161 | 130-160 |
| 6 | 164-167 | 163-166 |
| 7 | 170-180 | 169-179 |
| 8 | 195-211 | 194-210 |
| 9 | 233-236 | 233-236 |
| 10 | 275-304 | 273-302 |
| 11 | 307-312 | 305-310 |
| 12 | 351-401 | 342-392 |
| 13 | 451-461 | 439-449 |
| 14 | 499-515 | 485-501 |
| 15 | 551-559 | 539-547 |
| 16 | 573-579 | 561-567 |
| 17 | 582-631 | 570-619 |
| 18 | 644-670 | 629-655 |
| 19 | 694-755 | 675-736 |
| 20 | 760-783 | 741-764 |
| 21 | 789-802 | 770-783 |
| 22 | 827-844 | 803-820 |
Из таблицы 3 можно заметить, что вначале одинаково выровненные блоки совпадают как у MAFFT, так и у T-Coffee. Затем пошли различия. Список одинаково выровненных колонок вне блока (первое число — колонка в выравнивании MAFFT, второе — T-Coffee): 65-65, 98-98, 117-117, 191-190, 221-221, 412-403, 563-551, 573-561, 579-567, 638-623, 641-626, 668-669.
Построение выравнивания по совмещению структур и сравнение его с выравниванием программой MSA
Для анализа был выбран домен PF00030 (Beta/Gamma crystallin). Кристаллины представляют собой группу водорастворимых структурных белков, которые в высокой концентрации присутствуют в хрусталике и роговице глаза. Были выбраны белки c PDB ID 1AMM (из организма Bos taurus), 1HK0 (из Homo sapiens) и 1A45 (также из орагнизма Bos taurus). Мне было интересно сравнить два бычьих кристаллина с человеческим, поэтому в качестве референса был выбран 1HK0. Было проведено два множественных выравнивания: выравнивание по совмещению структур выбранных трех белков и с помощью программы MAFFT. Выравнивание по совмещению структур осуществлялось с помощью алгоритма TM-align на сайте PDB.
Проект Jalview с выравниванием по совмещению структур (первые три строчки) и с помощью программы MAFFT (последние три строчки). Сравнение выравниваний проводился так же с помощью программы, сделанной моими однокурсниками. По данным из таблицы 4, оба множественных выравнивания довольно хорошо совпадают и имеют большое количество одинаково выровненных колонок. Из изображения совмещения структур (рис. 1) и из множественного выравнивания по совмещению структур видно, что данные белки имеют высоко консервативную вторичную структуру и имеют очень много схожих участков, поэтому данные белки гомологичны по всей длине. Вариабельные участки наблюдаются в петлевых доменах белка. Из выравнивания, осуществленного программой MAFFT, можно сделать точно такой же вывод о гомологичности данных белков.
| Block | PDB Alignment | MAFFT |
|---|---|---|
| 1 | 1-82 | 1-82 |
| 2 | 87-116 | 87-116 |
| 3 | 122-175 | 121-174 |
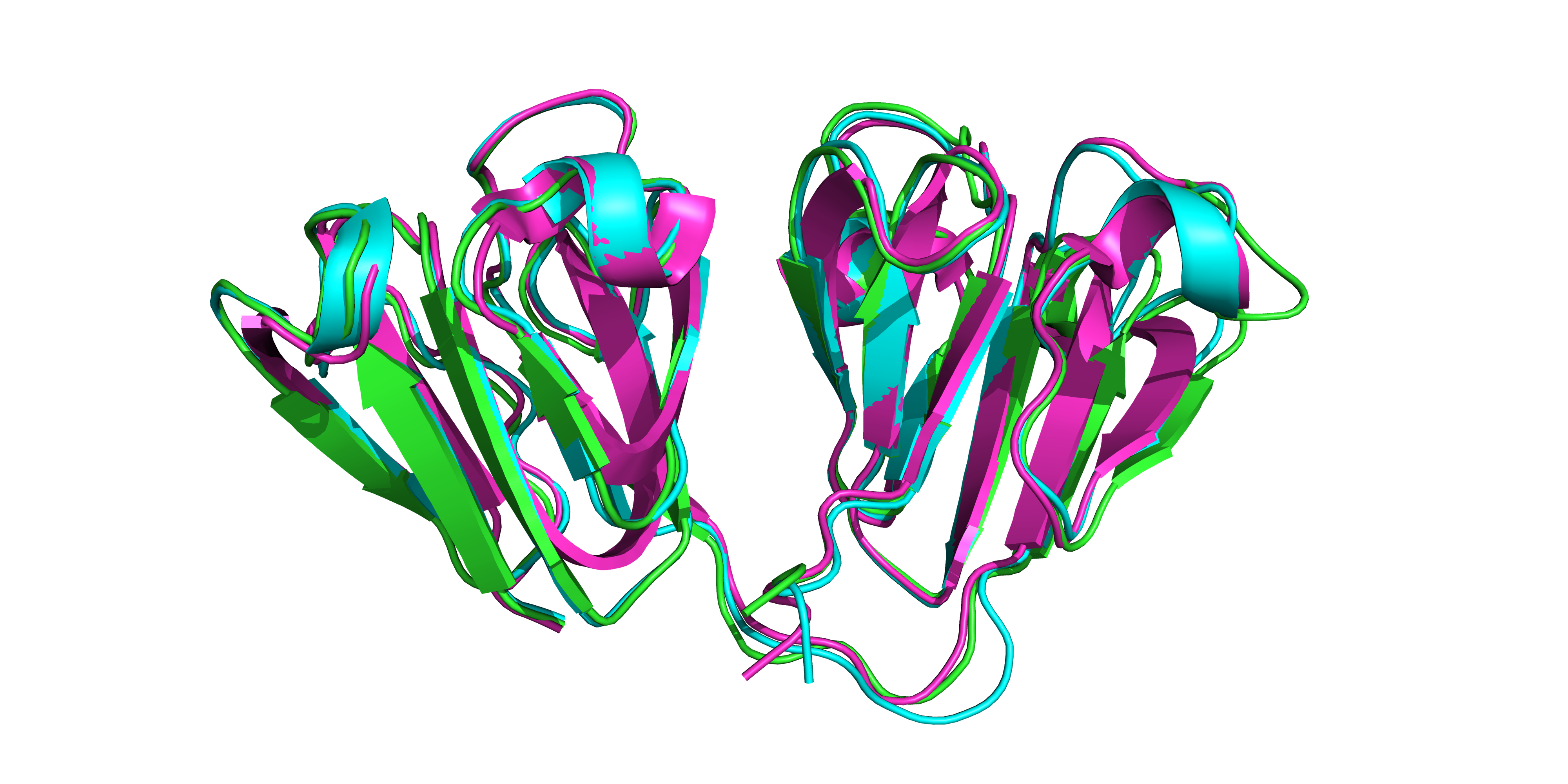
Краткое описание программы MUSCLE
Программа для проведения множественных выравниваний MUSCLE работает в три этапа, суть которых состоит в постепенном улучшении точности предсказанного выравнивания.
Первый этап заключается в проведении парных выравниваний между всеми последовательностями. Сначала программа оценивает сходство между всеми последовательностями, сравнивая короткие фрагменты (k-меры). На основе этих данных строится приблизительное дерево родства с помощью метода UPGMA. Затем выполняется предварительное, или черновое, выравнивание, где последовательности постепенно объединяются согласно этому дереву (от самых схожих к менее схожим).
Второй этап состоит в постройке второго, более улучшенного направляющего дерева. Теперь программа использует уже выровненные последовательности, чтобы более точно пересчитать расстояния между ними. Для оценки расстояния между ними испольуется так называемый метод Кимуры. Строится новое, более точное направляющее дерево. Выполняется повторное выравнивание, но только для тех частей дерева, где изменился порядок соединения последовательностей.
На третьем этапе программа анализирует полученное выравнивание и пытается его улучшить. Для этого она по очереди "разрезает" дерево на части и пробует перевыровнить эти части заново. Если новое выравнивание лучше (по специальной оценке), оно сохраняется. Этот процесс повторяется до тех пор, пока выравнивание не перестанет улучшаться.
Источник: Robert C. Edgar. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, Volume 32, Issue 5, 1 March 2004, Pages 1792–1797, https://doi.org/10.1093/nar/gkh340