1. Построение дерева по нуклеотидным последовательностям
AC записей EMBL и координаты генов рРНК:
AE016879 BACAN 29129-30635
AE015927 CLOTE 8715-10223 (compl.)
AE016830 ENTFA 248466-249987
AP008971 FINM2 197837-199361
CR954253 LACDA 45160-46720
AM406671 LACLM 511423-512971
AL591980 LISMO 96266-97811 (compl.)
AE015929 STAES 1598006-1599559 (compl.)
1. Для объединения всех последовательностей в единый файл воспользовался:
cat *.seq >> all.seq
2. Провел выравнивание программой muscle.
3. Далее использовал все шесть программ для получения деревьев. Таким образом, получил пять деревьев.
Ни одно дерево не совпало с правильным. Вообще, результаты данного подхода являются несколько менее качественными, по сравнению с "белковым" подходом.
И это кажется вполне логичным.
Во-первых, качество нуклеотидного выравнивания не самое высокое, так как не существует никаких функциональных
групп нуклеотидов, как у аминокислот, и замена одного нуклеотида на любой другой влечет за собой одинаковый штраф. Поэтому на этом этапе нужно уже учитывать
большую погрешность.
Во-вторых, в данном подходе не различается, является ли данная нуклеотидная замена (имеется в виду:
в одной последовательности по сравнению с другой) столь важной для разведения видов с эволюционной точки зрения. Ведь, например, мутация одного нуклеотида может даже не изменить аминокислотный остаток или же изменить, но на родственный.
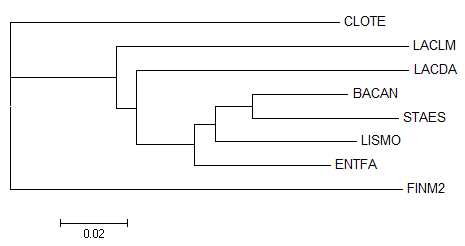
В качестве наиболее лучшего варианта я выбрал дерево из программы fneighbor.
Выдача fneighbor
| Правильное дерево
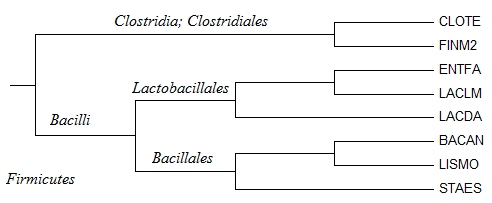 |
Если укоренить дерево на втором узле слева, то, в прнципе, так или иначе мы получим дерево, очень похожее на верное. Действительно, мы имеем кладу с Clostridia (CLOTE, FINM2), Bacillales (BACAN, STAES, LISMO), a Lactobacillales (ENTFA, LACLM, LACDA) хоть и нечетко, но отделяются от предыдущих. .
Вообще мне кажется, что крайне неправомерно называть полученное дерево "неправильным", ведь оно отражает эволюцию одной рибосомной РНК, никаких других требований к нему предъявлять нельзя.
А несоответствие "правильному" дереву - это уже проблема выбора объекта. Чтобы проанализировать эволюцию, нужно взять для анализа как можно больше белков и РНК.
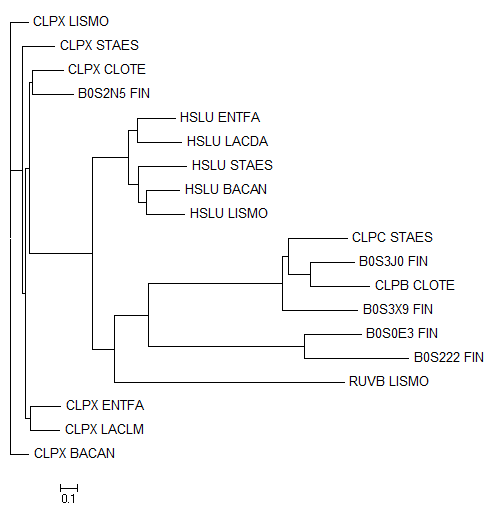
2. Построение и анализ дерева, содержащего паралоги
1. Использовал программы makeblastdb и blastp для поиска гомологов CLPX_BACSU. Всего 19 гомологов.
2. Получил последовательности благодаря сервису retrieve на сайте UniProt и выровнил их программой mafft.
3. Использовал программы fprotdist и fneighbor для построения дерева.
Паралогами являются:
B0S222_FINM2, B0S3J0_FINNM2, B0S3X9_FINM2 и B0S2N5_FINM2
CLPX_BACAN и HSLU_BACAN
Ортологами:
CLPX_STAES и CLPX_LISMO (если, конечно, разделение их общего предка на линии произошло в результате видообразования) и т.д.