Сигнал связывания CTCF
Поиск гомологов
CTCF - важнейший транскрипционный фактор, ремоделирующий хроматин посредством образования петель ДНК, связанных с ядерной ламиной [1]. В геноме человека было охарактеризовано более 30000 сайтов связывания CTCF [2]. Белок содержит 11 доменов ''цинковые пальцы'' и использует различные комбинации этих доменов для связывания с ДНК [3].
CTCF связывает консенсус CCACCAGGGGGCGC длиной в 14 оснований (Рис. 1) и допускает значительную вариабельность (этот мотив присутствует только в 80% экспериментально определённых сайтов связывания) [4]. Мотив высоко консервативен среди позвоночных животных [4].
А теперь вкратце:
A) Сигнал - последовательность ДНК из 20 пар оснований, среди которых самыми консервативными являются 14 букв CCACCAGGGGGCGC.
B) Сигнал адресован транкрипционному фактору CTCF
C) CTCF связывается с консенсусом и далее выполняет одну из своих функций: формирует петли (ремоделирование хроматина), блокирует распространение гетерохроматина (инсуляторная функция), блокирует взаимодействие промотор - энхансер и так далее (функции CTCF - это на данный момент довольно горячая тема в среде биоинформатиков, занимающихся хроматином).
D) Сигнал высокоэффективен. Отсутствие связывания может наблюдаться лишь при определённых условиях (например, при метилировании цитозина [5])
Построение матрицы PWM.
В данном задании я решил работать с консенсусом Козак, играющим важную роль в инициации трансляции у эукариот.
С кодом, использованным для этого задания, можно ознакомиться здесь. Код любезно предоставлен Селифоновым Игорем.
Скачав таблицу с человеческими генами, я случайным образом выудил из неё 100 последовательностей вида: 7 нуклетидов до ATG + ATG + 3 нуклеотида после ATG. При этом 40 из этих последовательностей были выбраны в качестве тренировочной выборки, а оставшие (60) - в качестве тестовой.
Далее я отобрал 60 последовательностей в материал негативного контроля, использовав для этого окружение нестартовых ATG-триплетов в геноме Sars-CoV-2 того же паттерна, что и предыдущие последовательности в train и test (7 нуклеотидов до ATG + ATG + 3 нуклеотида после ATG). Для этого использовалась разметка ATG-триплетов генома вируса.
После этого была создана позиционная весовая матрица. При этом при подсчёте логарифма отношения для устранения нулевых частот использовались pseudocounts, равные 0.1 для всех букв. Апостериорные частоты встречаемости оснований были рассчитаны в соответствии с GC-составом человеческого генома, равным 0.404
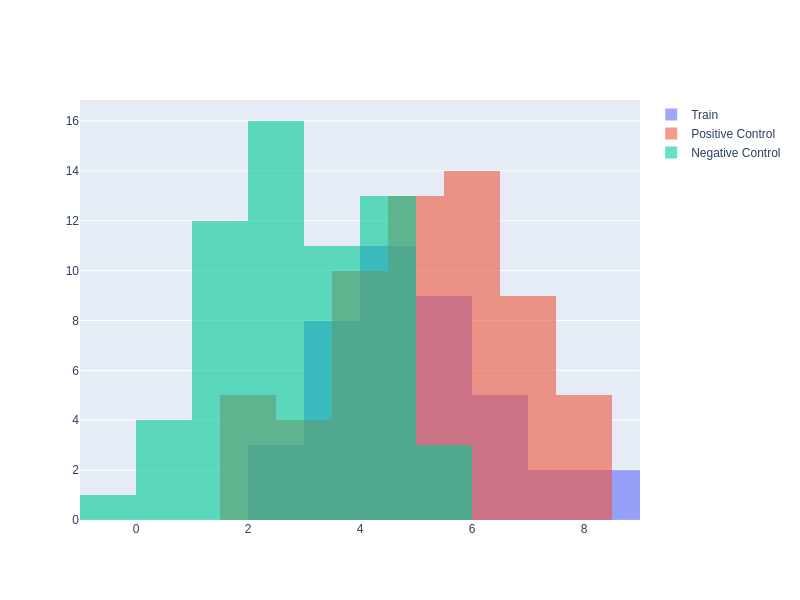
Далее был произведён подсчёт весов с помощью PWM для всех вышеперечисленных выборок: train, test (Positive Control) и Negative Control. По полученным данным была построена гистограмма весов (Рис. 2). Как и ожидалось, негативный контроль визуально сильно отличается по знечениям весов от train и test групп. Выберем порог весов, равный 4.0. Этот порог довольно удовлетворительно отделяет отрицательный контроль от положительного.
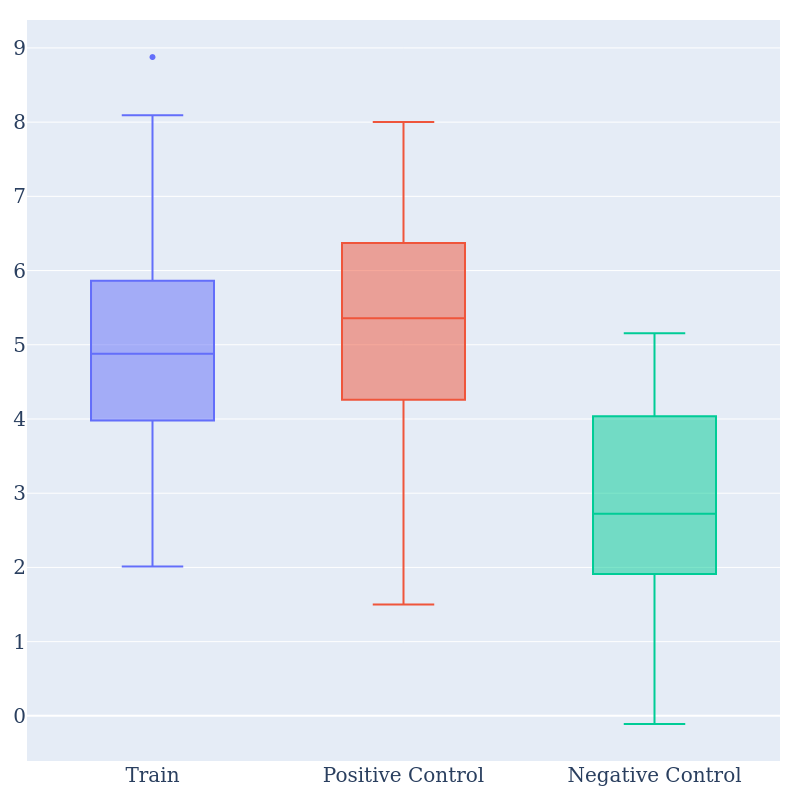
Также с помощью Plotly были построены ящики с усами, отображающими те же различия между тренировочным и тестовым наборами и отрицательным контролем (Рис. 3)
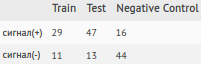
После этого я построил таблицу с количеством последовательностей, отобранных по вышеопределённому порогу (Таблица 2). Хотя из-за такого порога в группу (-)-сигнал попали некоторые последовательности из train (которые точно является сигналами), по большей части данный порог неплохо разделяет негативный и положительный контроли.
Построение матрицы IC и Logo консенсуса Козак.
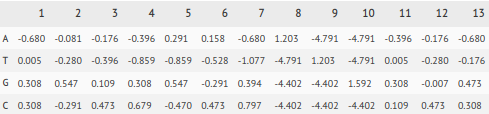
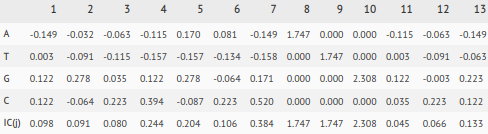
Далее на основе тренировочного набора была построена матрица IC (Таблица 3). При этом использовались те же данные, что и при построении матрицы PWM.
С помощью онлайн-ресурса WebLogo было получено Logo консенсуса Козак по тренировочной выборке (Рис. 3). В пятом положении хорошо выражен пурин (R), а в положениях 8-10 - стартовый триплет ATG.
Сайт GAATTC в геноме штама E. coli
В этом задании я скачал геном штама E. coli FORC_028, у которого GC-content равен 0.506.
Несколькими способами я посчитал количество 6-меров GAATTC. Их оказалось всего 805 на весь геном. При этом ожидаемое число соответствующих 6-меров я посчитал через произведение частот соответствующих оснований (с учётом GC-состава), помноженное на длину генома. В итоге ожидаемое число сайтов оказалось равным 1359.
Разница между полученными значениями равна 554. Для оценки статистической значимости я провёл тест Хи-квадрат, реализованный в пакете scipy.stats. H0 (нулевая гипотеза) заключается в том, что нет никакой статистически значимой разницы между ожидаемым и наблюдаемым значениями. При этом H1 (альтернативная гипотеза), соответственно, говорит о том, что различие между вышеупомянутыми значениями статистически значимо. Полученное p-value, равное около 1e-32, позволяет отвергать нулевую гипотезу и верить в альтернативную при почти сколь угодно малом пороге значимости.