ChIP-seq
Для работы нам были выданы файлы .fastq с одиночными прочтениями (ридами) illumina, полученными в результате ChIP-seq эксперимента. На первом этапе я сделала контроль качества прочтений с помощью программы FastQC. Я воспользовалась версией, установленной на сервере kodomo. В командной строке я написала:
fastqc chipseq_chunk46.fastq
После чего программа в той же папке выдала мне файл chipseq_chunk46_fastqc.html, содержащий визуализированный отчёт о работе, и архив chipseq_chunk46_fastqc.zip с каринками и текстовыми файлами. Качество прочтений было удовлетворительным, но усы мне показались слишком большими, поэтому следующим шагом я решила выполнить очистку чтений с помощью программы Trimmomatic. Я воспользовалась версией программы Trimmomatic, установленной на сервере kodomo. В командной строке я писала следующую команду:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chipseq_chunk46.fastq chipseq_chunk46_out.fastq TRAILING:50 MINLEN:36
Здесь опция TRAILING удаляет с конца чтения нуклеотиды с качеством ниже указанного, а опция MINLEN удаляет чтения с длиной меньше указанного числа. В результате было удалено 14,65% ридов, и я получила очищенный файл chipseq_chunk46_out.fastq, качество которого было представлено на странице chipseq_chunk46_out_fastqc.html
Теперь выполним сравнение чтений до и после очистки.
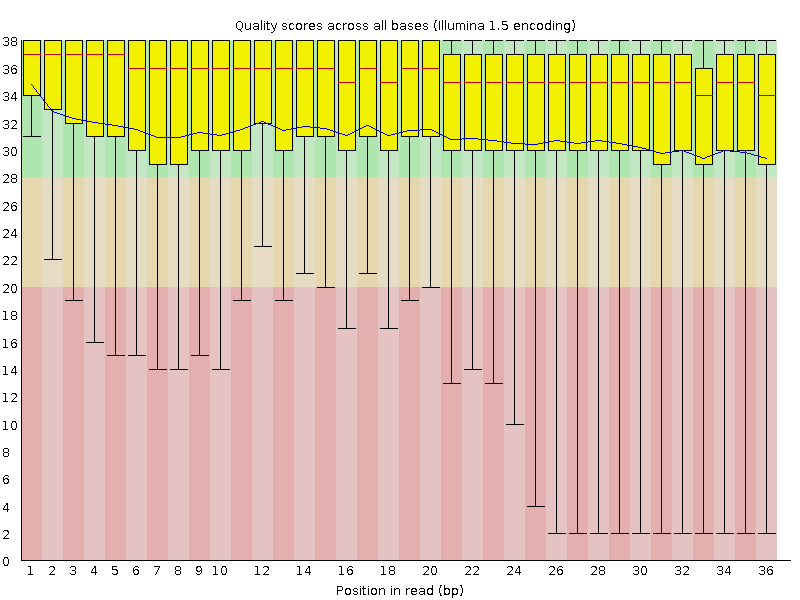
1) До очистки. Число чтений = 17258. Длина чтений = 36. Процен GC = 48%.
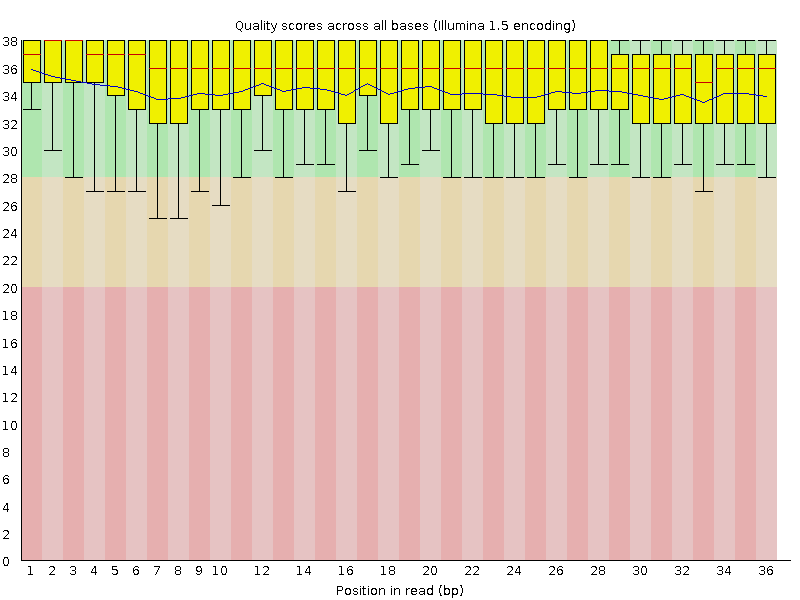
2) После очистки. Число чтений = 14730. Длина чтений = 36. Процен GC = 48%.
Затем я выполнила картирование прочтений на геном человека hg19 с помощью программы BWA. Для этого я выполнила команду:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk46_out.fastq > chipseq_chunk46.sam
Работа программы заняла 64 секунды, и на выходе я получила файл chipseq_chunk46.sam. Теперь мне нужно было перевести его в бинарный формат .bam. Для этого я воспользовалась следующей командой пакета samtools:
samtools view chipseq_chunk46.sam -bSo chipseq_chunk46.bam
Опция -b меняет формат файла на .bam, а опция -o указывает на название выходного файла. В результате я получила файл chipseq_chunk46.bam. Затем я отсортировала получившийся файл по координате начала чтения в референсе. Для этого я использовала команду:
samtools sort chipseq_chunk46.bam -T chip_temp -o chipseq_chunk46_sorted.bam
Опция -T позволяет записывать информацию во временные файлы, а не в stdout, а опция -o указывает на название выходного файла. В результате я получила отсортированный файл chipseq_chunk46_sorted.bam. Затем было необходимо его проиндексировать, для чего я воспользовалась командой:
samtools index chipseq_chunk46_sorted.bam
В результате я получила индексный файл chipseq_chunk46_sorted.bam.bai. Последней задачей по работе с пакетом samtools было выяснить, сколько чтений откартировалось на геном. Для этого я воспользовалась командой:
samtools idxstats chipseq_chunk46_sorted.bam > out.txt
В результате я получила файл out.txt в котором указаны название последовательности, длина последовательности и число картированных на нее чтений. Получается, что из 14730 чтений 12826 (87%) были откартированы на 11 хромосому. Причем на геном откартировались все прочтения. Поэтому я считаю, что мне для анализа были предложены прочтения именно с 11 хромосомы.
Для поиска пиков я воспользовалась версией программы MACS, установленной на сервере kodomo. В командной строке я написала:
macs2 callpeak -n chunk46 -t chipseq_chunk46_sorted.bam --nomodel
В результате я получила файлы chunk46_peaks.xls, chunk46_peaks.narrowPeak и chunk46_summits.bed с информацией о найденных пиках, представленной в таблице 1. Всего было найдено 12 пиков,причем один из них не относится к 11 хромосоме. В дальнейшем я решила не исследовать его.
| Таблица 1. Информация о найденных пиках | ||||||||
| № | Хромосома | Начало | Конец | Длина | Вершина | Расстояние от начала пика до его вершины |
pileup | -log10(p-value) |
| 1 | GL949744.1 | 170397 | 170630 | 234 | 170536 | 139 | 19.00 | 18.63553 |
| 2 | chr11 | 67769619 | 67769833 | 215 | 67769761 | 142 | 20.00 | 18.68741 |
| 3 | chr11 | 67844008 | 67844262 | 255 | 67844077 | 69 | 17.00 | 14.75298 |
| 4 | chr11 | 67905323 | 67905522 | 200 | 67905492 | 169 | 18.00 | 15.94685 |
| 5 | chr11 | 67914504 | 67914944 | 441 | 67914703 | 199 | 39.00 | 30.20449 |
| 6 | chr11 | 68147878 | 68148282 | 405 | 68148068 | 190 | 36.00 | 30.99165 |
| 7 | chr11 | 68235834 | 68236084 | 251 | 68235918 | 84 | 24.00 | 18.20288 |
| 8 | chr11 | 68612957 | 68613239 | 283 | 68613096 | 139 | 33.00 | 29.79350 |
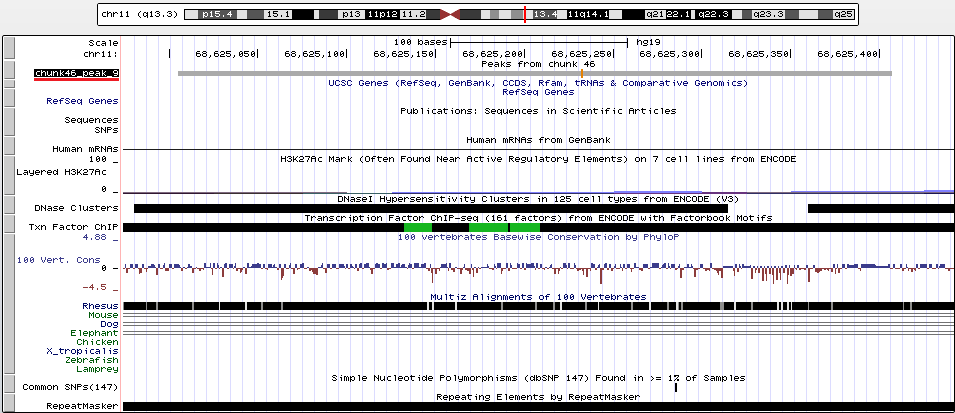
| 9 | chr11 | 68625006 | 68625407 | 402 | 68625233 | 227 | 47.00 | 43.37504 |
| 10 | chr11 | 69383055 | 69383254 | 200 | 69383153 | 98 | 16.00 | 14.55495 |
| 11 | chr11 | 69469682 | 69470047 | 366 | 69469814 | 132 | 25.00 | 20.60395 |
| 12 | chr11 | 69816533 | 69816732 | 200 | 69816614 | 81 | 15.00 | 14.71966 |
Отметим, что чем выше показатель -log10(pvalue), тем ниже соответствующее значение p-value. Cледовательно, cамым достоверным является пик 9, а наименее достоверным - пик 10.
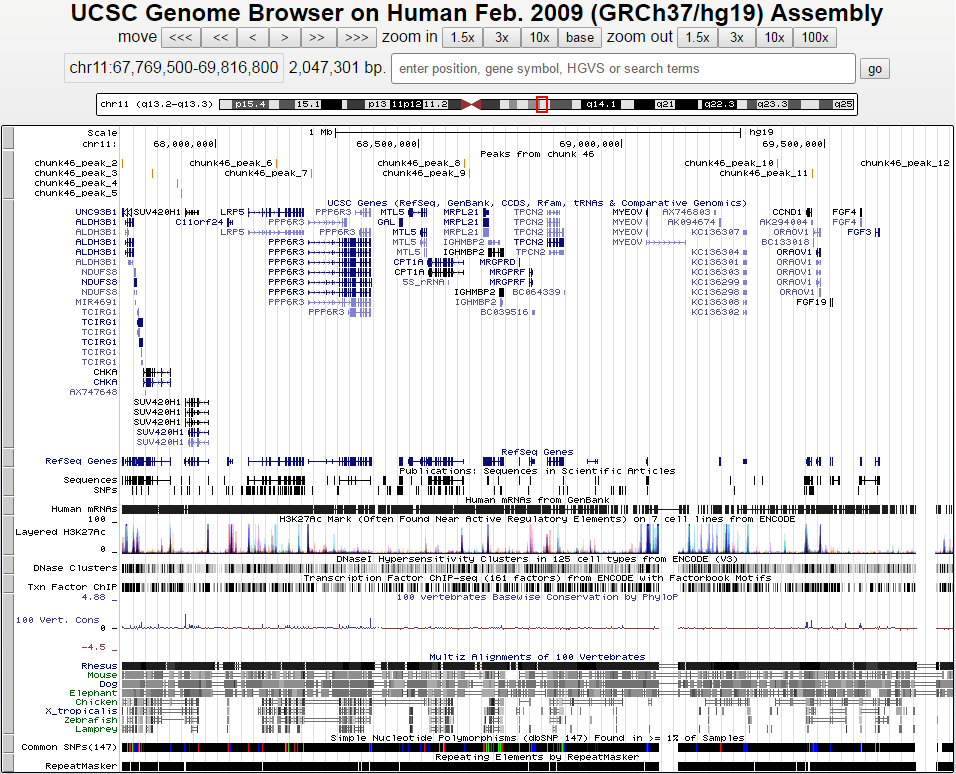
Визуализация пиков была проведена с помощью сайта UCSC Genome Browser. На вход подавался файл chunk46_peaks.narrowPeak, в начало которого были дописаны две строчки:
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 46" browser position chr11:67769500-69816800
Ниже представлено изображение пиков, полученное с помощью UCSC Genome Browser
Далее я решила подробнее рассмотреть пики 7 и 9. Ниже представлено увеличенное изображение пика 7 (название подчеркнуто красной линией слева). Расположение пика приходится на интрон гена PPP6R3, кодирующего регулятугную субъединицу 3 серин/треонин-белковой фосфотазы 6.
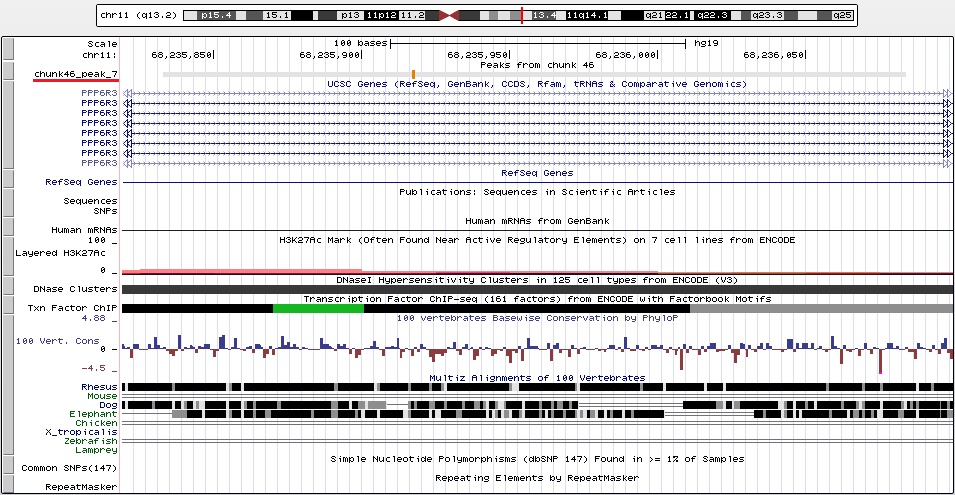
Ниже представлено увеличенное изображение пика 9 (название подчеркнуто красной линией слева). Расположение пика не приходится ни на один ген.