Реконструкция эволюции доменной архитектуры
Выбор семейства доменов
При выборе семейства доменов необходимо было учитывать следующие пункты:
- В семействе должно быть не более 10 000 последовательностей
- В семействе представлены последовательности не более чем из 1 200 видов
- Домены семейства входят как минимум в две разные доменные архитектуры, представленные как минимум парой десятков последовательностей (>20) каждая
- Хотя бы для одной последовательности известна 3D структура белка (домена)
Поиск семейства доменов проводился в базе данных Pfam. Под указанные выше требования подошел домен Usher (PF00577) из мембранного белка FIMD_ECOLI (UniProt), участвующего в сборке фимбриальных субъединиц FimA и их экспорте через наружную мембрану. Ранее этот белок рассматривался в практикуме с идентификатором 3OHN в базе данных OPM. Ниже представлено изображение с краткой информацией о домене Usher

Затем я с помощью программы JalView скачала выравнивание последовательностей выбранного семейства из Pfam (Full), указав AC = PF00577. Раскрасила выравнивание по типу ClustalX. Указывать окраску по консервативности не имело смысла, так как в таком случае ни один аминокислотный остаток не был бы окрашен. В полученном выравнивании было достаточно много гэпов и мало консервативных колонок. Также я добавила к файлу с проектом 3D структуру 4J3O самой верхней последовательности белка FIMD_ECOLI (UniProt). Также полученное выравнивание доступно в fasta формате.
Выбор архитектур
Для семейства доменов Usher (PF00577) мне предстояло выбрать 2-3 архитектуры, соответствующие требованиям:
- Простота: одно-, двух-, или трех-доменные архитектуры
- Должно быть не менее 20 последовательностей с данной архитектурой, на считая фрагментов
Данным требованиям удовлетворяют архитектуры {PapC_N, Usher, PapC_C} (641 последовательность) и {PapC_N, Usher} (90 последовательностей).
Ниже приведено изображение архитектуры {PapC_N, Usher, PapC_C} (641 последовательность). Далее для удобства буду называть ее "архитектура A". Синим цветом обозначен домен PapC_C. Прямоугольники с закругленными краями обозначают домены, которые полностью встречаются в данных последовательностях.

Ниже приведено изображение архитектуры {PapC_N, Usher} (90 последовательностей). Далее для удобства буду называть ее "архитектура B". Прямоугольники с рваными краями обозначают домены, которые частично (отрывком) встречаются в данных последовательностях.

Далее мне нужно было получить таблицу с информацией об архитектуре всех последовательностей, содержащих домен Usher (PF00577). Для этого я сначала воспользовалась скриптом swisspfam_to_xls.py, чтобы отобрать все последовательности данного домена в таблицу для Excel из файла /srv/databases/pfam/swisspfam.gz, лежащего на сервере kodomo:
python swisspfam_to_xls.py -z swisspfam.gz -p PF00577 -o archs.xls
Опция -z разархивирует файл swisspfam.gz. Опция -p нужна для указания AC конкретного домена. Опция -o задает имя выходного файла. В результате я получила файл archs.xls, в котором перечислены идентификаторы последовательностей, их длина, описание, а также AC всех доменов и их координаты.
Далее, основываясь на полученной таблице, я составила сводную таблицу, где по строкам указаны AC последовательностей, а по столбцам - AC доменнов.
Затем мне нужно было в список последовательностей добавить колонки с информацией о таксономической принадлежностью. Поэтому сначала для всех последовательностей я скачала их полные записи из базы данных UniProt:
UniProt -> Retrieve/ID mapping -> upload file -> Go -> Download -> Download all -> Format: Text
Далее я с помощью скрипта uniprot_to_taxonomy.py получила нужную таксономию:
python uniprot-to-taxonomy.py -i flat.txt -o tax.xls
Данный скрипт преобразует информацию о таксономии из записей Uniprot (опция -i) в таблицу Excel (опция -o). В результате я получила файл tax.xls, информацию из которого я перенесла в основную таблицу.
На последнем шаге я добавила в таблицу колонку с длиной домена Usher из каждой последовательности.
В результате я получила таблицу final.xlsx.
Выбор таксона и подтаксонов
Как видно из финальной таблицы, большинство последовательностей принадлежит домену (доминиону) бактерий (Bacteria), типу протеобактерий (Proteobacteria). Далее я посмотрела какое распределение последовательностей из организмов протеобактерий по классам. Сравнение приведено в таблице 1.
| Таблица 1. Количество последовательностей из различных классов протеобактерий | |||
| Класс | Количество последовательностей |
Последовательности с архитектурой А |
Последовательности с архитектурой B |
| Gammaproteobacteria | 847 | 545 | 78 |
| Betaproteobacteria | 155 | 80 | 7 |
| Alphaproteobacteria | 85 | 0 | 0 |
| Deltaproteobacteria | 16 | 0 | 0 |
| Acidithiobacillia | 1 | 0 | 0 |
Требовалось выбрать такие подтаксоны, чтобы в них было не менее 20 последовательностей с каждой из архитектур. Вопреки данному требованию я решила остановиться на классах Gammaproteobacteria и Betaproteobacteria. В дальнейшем я буду использовать краткие обозначения (таблица 2).
| Таблица 2. Краткие обозначения | ||
| Класс | архитектура А | архитектура B |
| Gammaproteobacteria | GA | GB |
| Betaproteobacteria | BA | BB |
Выбор представителей архитектур
В основной таблице final.xlsx я добавила колонку (selected) для отметки выбранных мною последовательностей (помечены звездочкой, *). Нужно было взять по 20 или больше последовательностей из каждой архитектуры рассматриваемых таксонов. Причем они должны иметь примерно одинаковую длину. Также я добавила к выборке последовательность с известной 3D структурой ( FIMD_ECOLI, P30130).
Чтобы в выравнивании оставить только отобранные последовательности я воспользоваласть скриптом filter-alignment.py, предварительно подготовив файл ids.txt c AC выбранных последовательностей:
python filter-alignment.py -i pfam.mfa -m ids.txt -o pfam_selected.fasta -a '_'
Опция -i нужна для указания входного файла с выравниванием в fasta формате, опция -m нужна, чтобы задать файл с идентификаторами или AC последовательностей (должны совпадать со входным файлом), опция -o задает файл вывода, и опция -a позволяет задать символ разделителя. В результате из 76 последовательностей программа собрала только 71, так как некоторые последовательности были названы по-другому. Поэтому недостоющие последовательности я добавила вручную.
Затем я подкорректировала имена последовательностей, указав короткие обозначения (таблица 2) в начале, и с помощью программы JalView (Edit -> Remove Empty columns) убрала из выравнивания пустые колонки. В результате я получила файл с выравниванием pfam_selected.mfa. Далее в программе JalView я разбила последовательности на две группы (A и B) в зависимости от принадлежности к той или иной архитектуре. Раскрасила каждую группу по типу ClustalX с порогом консервации 7%. Отсортировала последовательности по группам. Также я удалила из выравнивания явные фрагменты, N- и C-концевые невыровненные участки и последовательности, которые по всей длине выровнены неправильно. Результат сохранен в виде проекта.
В целом можно сказать, что в обеих группах можно выделить более-менее консервативные участки (блоки). Однако, в группе, соответствующей архитектуре А консервативных позиций гораздо больше, чем в группе, соответствующей архитектуре B. Возможно, это связано с немногочисленностью выборки последовательностей из последней группы. Тем не менее, я считаю, что качество получившегося выравнивания удовлетворительно для построения филогенетического дерева домена.
Построение филогенетического дерева домена
Напомню еще раз, что в таблице 2 приведена расшифровка кратких обозначений, используемых в именах последовательностей. По выравниванию из проекта JalView я построила филогенетическое дерево (ниже приведено изображение) методом Neighbour Joining в программе MEGA. Для подтверждения достоверности ветвей применялся Bootstrap тест со 100 репликами.
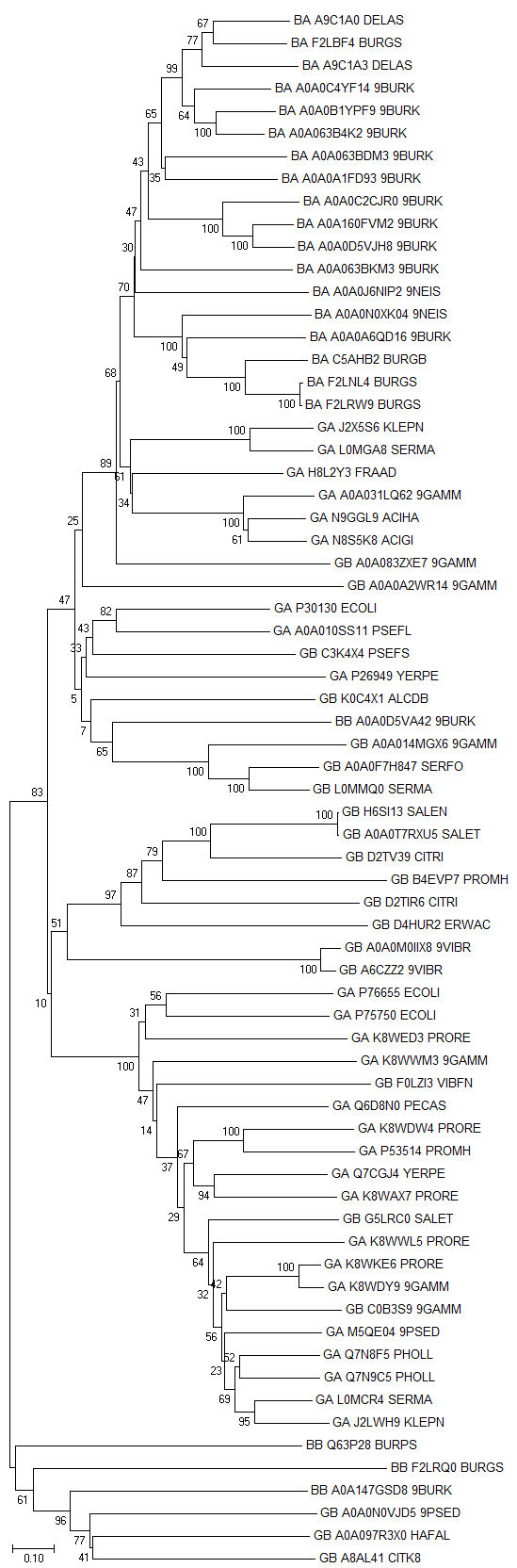
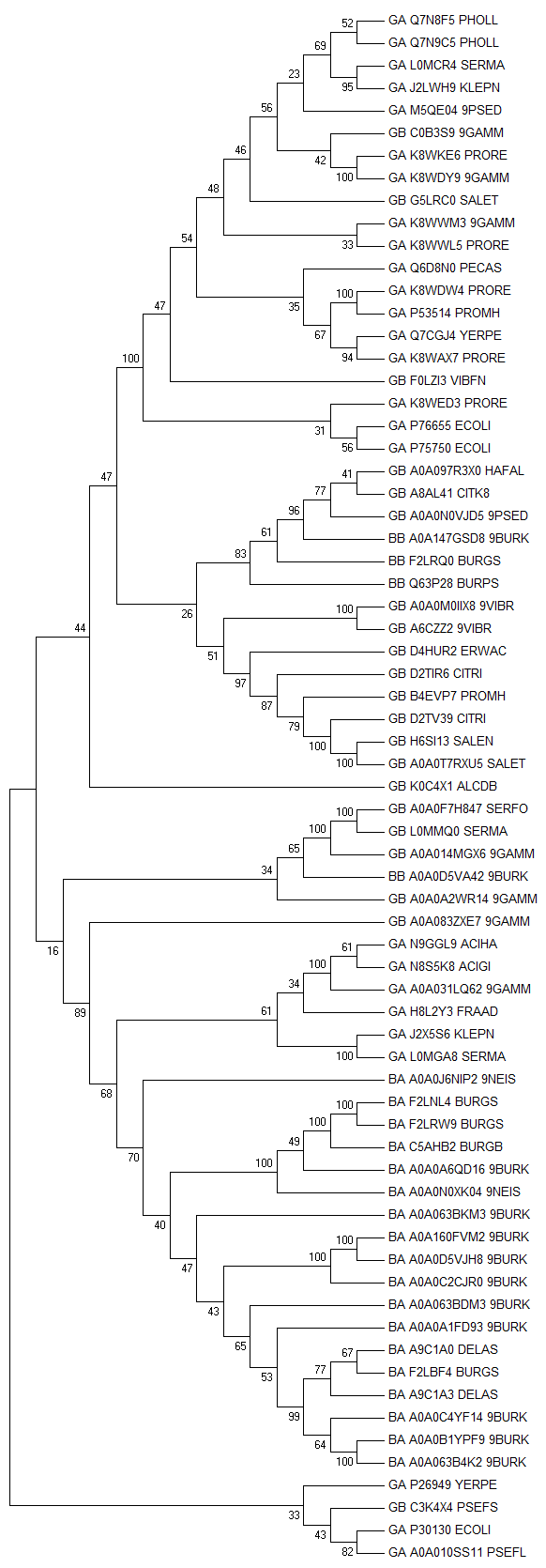
В файле NJ.nwk содержится скобочная структура дерева с указанием результатов бутстрэп-анализа. Ниже представлено изображение дерева, укорененного в среднюю точку, полученное с помощью программы ITOL. На ветвях подписаны результаты бутстрэп-теста. Синим цветом выделены названия последовательностей с архитектурой A, красным цветом - последовательности с архитектурой B. Фиолетовым цветом выделены клады, относящиеся к классу Gammaproteobacteria, зеленым цветом - к классу Betaproteobacteria.
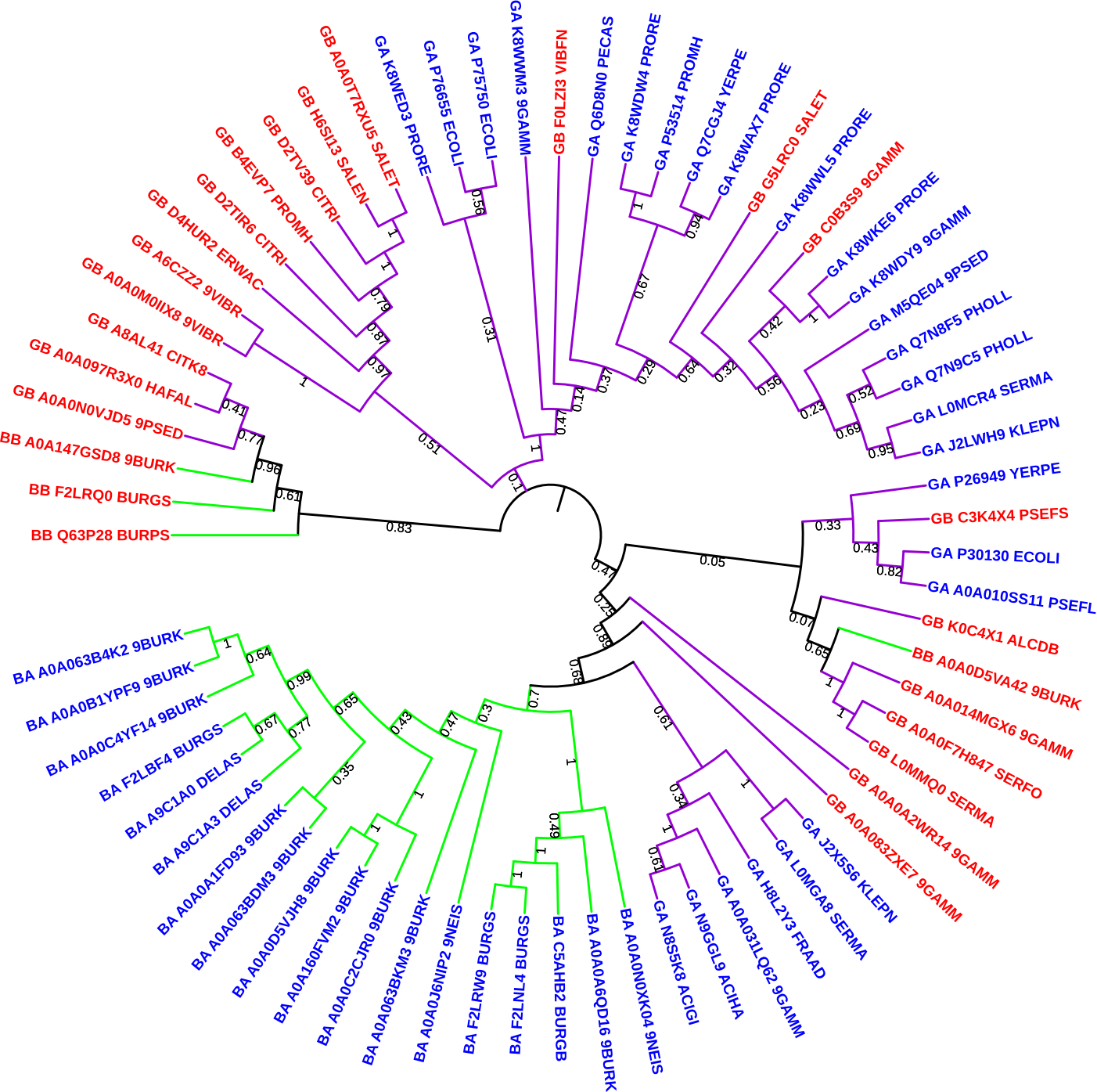
Если учитывать различие доменных архитектур A и B, то можно предположить, что белок с архитектурой А появился намного раньше, а затем в процессе эволюции в некоторых таксонах закрепилась его урезанная копия с архитектурой B. Интересно отметить, что в классе Betaproteobacteria для большинства видов оказалась более выгодной А-форма.