Совмещение структур
Отбор структурных гомологов
С помощью PDBeFold я получила перечень структурных гомологов белка пероксидазы хрена (PDB ID = 1KZM). У этого белка в PDB файле аннотировано NSSE = 18 элементов вторичной структуры. Из списка гомологов я выбрала следующие находки с 0.8 Å ≤ RMSD ≤ 2.5 Å и длиной выравнивания (Nalign) более 50 % от длины белка (Nquery = 308).
| Таблица 1. Структурные гомологи | ||||||
| Название | PDB ID | RMSD | Q-score | Nalign | NSSE | Nalign/Nquery |
| Arabidopsis Thaliana peroxidase A2 | 1QO4:A | 0.82 Å | 0.91 | 303 | 18 | 98.4 % |
| Raphanus Sayivus anionic peroxidase | 4A5G:A | 0.93 Å | 0.89 | 302 | 17 | 98.1 % |
| Arabidopsis Thaliana peroxidase N | 1QGJ:A | 0.97 Å | 0.85 | 294 | 17 | 95.5 % |
| Peanut peroxidase | 1SCH:A | 1.15 Å | 0.82 | 291 | 16 | 94.5 % |
Совмещение структуры HRP с его 4 структурными гомологами
Далее на странице находок я отметила представленныве выше структуры и с помощью кнопки "Submit for Multiple Alignment" получила множественное выравнивание malign.fasta со следующими характеристиками:
- Число выровненных остатков = 286.
- Число выровненных элементов вторичной структуры = 15.
- Общее RMSD = 0.9427 Å.
- Общий Q-score = 0.8248.
Ниже представлено изображение множественного структурного выравнивания.

Далее я получила файлы с совмещенными структурами ma.rasmol и ma.pdb. И с помощью PyMOL визуализировала последний PDB файл и получила следующее изображение совмещения выровненных структур.
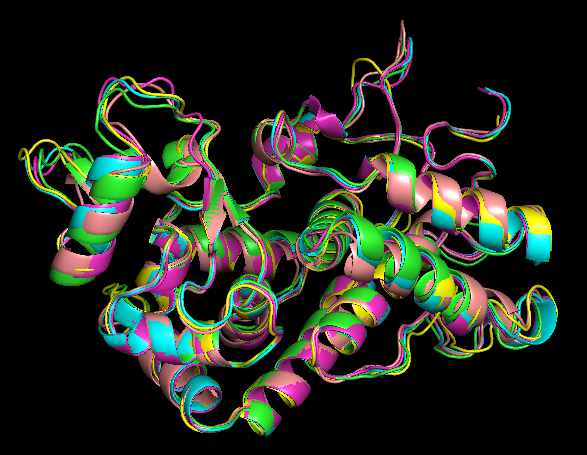
В целом, по картинке видно, что найденные гомологичные структуры хорошо накладываются друг на друга даже в области петель.
Следом было решено сравнить выравнивание последовательностей по структуре с выравниванием тех же последовательностей, построенным по алгоритму Muscle. Для визуализации использовалась программа Jalview.
Bыравнивание последовательностей по структуре.

Bыравнивание последовательностей по алгоритму Muscle.

В целом, оба выравнивания практически совпадают, за искючением позиций: 137-138, 159-160, 185-188, 254-261, 292-296 (нумерация в соответствии с выравниванием по структуре). Для позиций 137-138 я бы сделала выбор в пользу структурного выравнивания. Оно кажется более достоверным здесь, хотя само различие незначительно - позиция одного остатка в одной цепи. Аналогично, в случае позиций 159-160. Напротив, для позиций 185-188 более правильным кажется вариант, представленный в выравнивании по последовательностям, так как в этом случае будет больше число консервативных колонок, а также будет учитано, что Phe и Tyr структурно схожие остатки и замена одного на другой по позиции 186 весьма вероятна, как и вероятна эволюционная замена Leu на Ile (позиция 185). Для оставшихся двух областей я так и не определилась, какой вариант считать более достоверным. Думаю, для более однозначных выводов необходимо аналогичным образом построить выравнивания большего числа близкородственных белков.