Занятие 10. Докинг низкомолекулярных лигандов в структуру белка.
Цель занятия - ознакомится с возможностями докинга низкомолекулярного лиганда в структуру белка с помощью пакетов
Autodock Vina и Autodock tools.
Работу будем проводить с белком лизоцимом из зеленой морской черепахи (LYSC_CHEMY),
структура
которого была построена на основе гомологичного моделирования на прошлом практикуме.
- Программе Autodock Vina для докинга необходимы специально форматированные файлы pdb c зарядами и указанием торсионных углов.
Для начала попробуем провести докинг одного из мономеров сахара (NAG) из прошлого занятия.
В банке PDB найдем SMILES аннотацию для NAG: nag.smi.
- C помощью obgen построим 3D структуру этого сахара в pdb формате:
obgen nag.smi > nag.mol
babel -imol nag.mol -opdb nag.pdb
Выходной файл: nag.pdb.
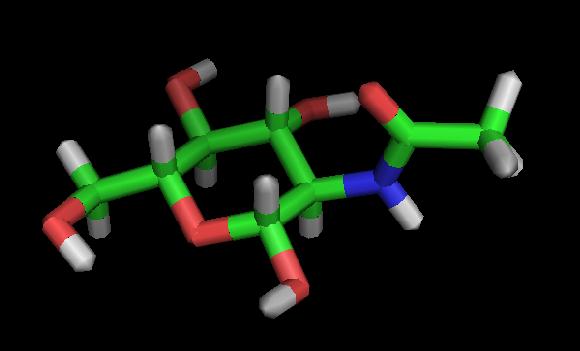
- С помощью скрипта prepare_ligand4.py из пакета Autodock tools создадим pdbqt-файл лиганда NAG:
prepare_ligand4.py -l nag.pdb
Выходной файл: nag.pdbqt.
- С помощью скрипта prepare_receptor4.py из пакета Autodock tools создадим pdbqt файл белка LYSC_CHEMY:
prepare_receptor4.py -r lysc_3.pdb
Выходной файл: lysc_3.pdbqt.
- Итак, у нас есть входные файлы. Теперь надо создать файл с параметрами докинга vina.cfg.
Для докинга необходимо указать область
структуры белка в которой будет происходить поиск места для связывания.
Удобно его задать как куб с неким центором. Координаты центра определим из
модели комплекса, построенной на прошлом занятии.
Для этого выберем какой-нибудьт атом сахара, аходящийся в центре сайта связывания,
и извлечем из текста его координаты:
HETATM 1054 N2B NAG B 131 40.992 42.120 26.552 1.00182.27 N
Построим файл vina.cfg с примерно таким содержанием:
center_x=40.992
center_y=42.120
center_z=26.552
size_x = 25
size_y = 25
size_z = 25
num_modes = 20
- Проведем первый докинг:
vina --config vina.cfg --receptor lysc_3.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt
--log nag_prot.log
Выходные файлы: nag_prot.pdbqt,
nag_prot.log.
- Просмотрим файл nag_prot.log и запишем энергии 3ёх лучших расположений и геометрическую разницу между ними:
| Расположение |
Энергия (ккал/моль) |
Геометрическая разница с лучшей моделью (rmsd l.b.) |
Геометрическая разница с лучшей моделью (rmsd u.b.) |
| 1 |
-5.6 |
0.000 |
0.000 |
| 2 |
-5.6 |
2.707 |
4.432 |
| 3 |
-5.2 |
1.934 |
5.027 |
Файлы nag_prot.pdbqt и lysc_3.pdbqt были загружены в PyMOL. Все состояния на одной картинке изображены ниже:
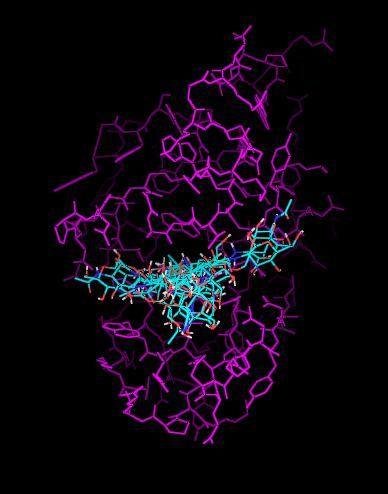
Из картинки видно, что лиганд свободно перемещается внутри центра связывания белка.
Возможно, это происходит из-за того, что мы докируем не весь комплексный лиганд, состоящий из трех сахарных остатков,
а только одну его часть (один сахар).
- Теперь проведём докинг, рассматривая подвижность некоторых боковых радикалов белка.
Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты, которые использовали
в прошлом задании для позиционирования лиганда.
prepare_flexreceptor4.py -r lysc_3.pdbqt -s ASP102_ASN104_VAL110
и проведём докинг:
vina --config vina.cfg --receptor lysc_3_rigid.pdbqt --flex lysc_3_flex.pdbqt
--ligand nag.pdbqt --out nag_prot_flex.pdbqt --log nag_prot_flex.log
- Просмотрим файл nag_prot_flex.log и запишем энергии 3ёх лучших расположений и геометрическую разницу между ними:
| Расположение |
Энергия (ккал/моль) |
Геометрическая разница с лучшей моделью (rmsd l.b.) |
Геометрическая разница с лучшей моделью (rmsd u.b.) |
| 1 |
-5.3 |
0.000 |
0.000 |
| 2 |
-5.1 |
1.746 |
4.026 |
| 3 |
-5.0 |
1.548 |
1.907 |
Докинг с подвижными радикалами считаются немного дольше, чем
докинг без подвижных радикалов.
Файлы nag_prot_flex.pdbqt и lysc_3_rigid.pdbqt были загружены в PyMOL. Все состояния на одной картинке изображены ниже:
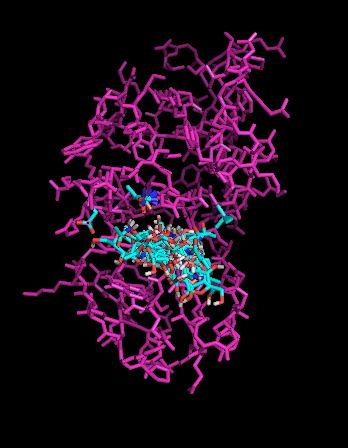
В данном случае свое положение меняет не только лиганд, но и три выбранных аминокислоты белка: Asp102, Asn104, Val110.
Из рисунка видно, что наиболее изменяется конформация у Asn104. Положение лиганда также влияет и на конформацию Val110, а вот на Asp102 влияение не очень сильно, остаток лишь чуть-чуть колеблется (на рисунке не заметно).
Мне кажется, что с биологической точки зрения разумнее делать подвижный докинг, ведь белок не является неподвижной жесткой структурой.
- В моделировании мы получили следующую картину:
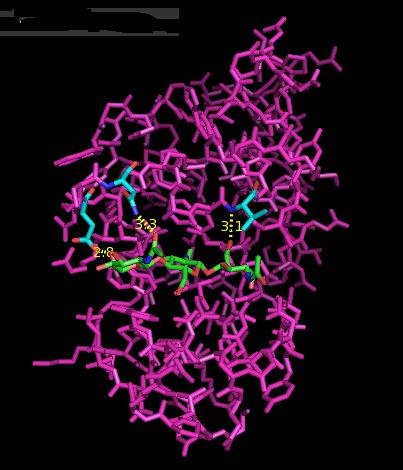
К сожалению, докинг не смог расположить лиганд наиболее близким образом к тому положению, что он занимал в результате
моделирования (например, водородная связь между 102 остатком и лигандом так и не образовалась).
- NAG содержит в себе СH3C(=O)NH группу. Создадим 3 лиганда, где метильный радикал этой группы будет заменён на OH,
NH2,H: nag_oh.smi,
nag_nh2.smi, nag_h.smi.
PDB-файлы: nag_oh.pdb, nag_nh2.pdb,
nag_h.pdb.
C помощью скрипта prepare_ligand4.py были получены pdbqt-файлы: nag_oh.pdbqt,
nag_nh2.pdbqt, nag_h.pdbqt.
Затем для каждого лиганда провели обычный докинг.
Результаты для первого лиганда: nag_prot_oh.log,
nag_prot_oh.pdbqt.
Энергия 3ёх лучших расположений и геометрическая разница между ними:
| Расположение |
Энергия (ккал/моль) |
Геометрическая разница с лучшей моделью (rmsd l.b.) |
Геометрическая разница с лучшей моделью (rmsd u.b.) |
| 1 |
-5.0 |
0.000 |
0.000 |
| 2 |
-4.9 |
6.564 |
8.139 |
| 3 |
-4.8 |
1.005 |
3.723 |
Результаты для второго лиганда: nag_prot_nh2.log,
nag_prot_nh2.pdbqt.
Энергия 3ёх лучших расположений и геометрическая разница между ними:
| Расположение |
Энергия (ккал/моль) |
Геометрическая разница с лучшей моделью (rmsd l.b.) |
Геометрическая разница с лучшей моделью (rmsd u.b.) |
| 1 |
-4.9 |
0.000 |
0.000 |
| 2 |
-4.8 |
7.011 |
8.185 |
| 3 |
-4.7 |
6.627 |
8.198 |
Результаты для третьего лиганда: nag_prot_h.log,
nag_prot_h.pdbqt.
Энергия 3ёх лучших расположений и геометрическая разница между ними:
| Расположение |
Энергия (ккал/моль) |
Геометрическая разница с лучшей моделью (rmsd l.b.) |
Геометрическая разница с лучшей моделью (rmsd u.b.) |
| 1 |
-4.7 |
0.000 |
0.000 |
| 2 |
-4.7 |
2.297 |
3.018 |
| 3 |
-4.6 |
1.223 |
3.578 |
|
|