Пакет Pftools
Занятие 12.
1. Готовим входной файл в формате msf
В качестве выравнивания берем выравнивание паттернов моего семейства из прошлого занятия, сохраняем в msf формате. Далее готовим входной файл, для этого используем программу noreturn, запускаем ее и вбиваем имя входного и выходного файла. Эта программа меняет признаки конца строки на UNIX-вые симфолы.
2. Рассчитываем веса строк моего выравнивания программой pfw
Для этого использовали команду: pfw -in Gene3.msf -out Gene3.msf
Полученный файл отличается от входного тем, что веса строк не равны 1.
3. Создаем профиль программой pfmake
Команда: pfmake Gene3.msf /usr/share/pftools23/blosum62.cmp > all.prf
4. Подготовка файла с последовательностями в fasta-формате, в которых будет проводиться поиск
Команда: seqret sw-org:bacteria bacteria.fasta
5. Нормируем профиль
Процедура нормировки меняет формулу пересчёта обычной суммы весов в нормированный вес, это облегчает установку порога для данного профиля. Для нормировки прежде всего нужно сгенерировать случайный банк того же размера, что мой.
Это делается с помошью команды: shuffleseq sw-org:bacteria shuffled.fasta
Теперь проводим "фальшивый поиск", чтобы получить типичные значения веса профиля на случайных последовательностях.
Это делается с помошью команды: pfsearch -C0.0 -f my.prf shuffled.fasta | sort -n > scores.txt
Затем делаем нормировку профиля программой pfscale с помощью команды: pfscale scores.txt my.prf > scaled.prf
Получили файл scaled.prf, он отличается от не нормарованного файла all.prf данными в строчке:
MA /NORMALIZATION: MODE=1; FUNCTION=LINEAR; R1=NaN; R2=NaN; TEXT='-LogE'; - в нормированном файле;
MA /NORMALIZATION: MODE=1; FUNCTION=LINEAR; R1=0.0000000; R2=0.0100000; TEXT='No_units'; - в не нормарованном файле.
6. Поиск по профилю
Пользуемся командой: pfsearch -C1.7 -f all.prf bacteria.fasta > my.pfsearch
Взяли порог 1.7, т.к. с порогом 1.0 обнаруживается свыше 120 тыс. находок. Сейчас 11768 находок.
7. Анализ результатов
Построили ROC-кривую. См. лист "roc" в файле EXCEL
Или на картинке:

Для нее характеристики:
Число верных находок ("True positive hits", TP): 55;
Число ложных находок ("False positive hits", FP): 11354;
Число ненайденных белков подсемейства (ложноотрицательных результатов, "False negatives", FN): 0;
Чувствительность TP/(TP+FN): 100%;
Селективность TP/(TP+FP): 0,48%.
Если взять порог 8,36, то параметры несколько изменятся. (См. лист "мойпат" в файле EXCEL)
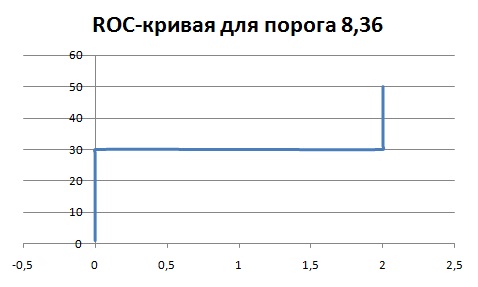
Для нее характеристики:
Число верных находок ("True positive hits", TP): 50;
Число ложных находок ("False positive hits", FP): 2;
Число ненайденных белков подсемейства (ложноотрицательных результатов, "False negatives", FN): 5;
Чувствительность TP/(TP+FN): 90,90%;
Селективность TP/(TP+FP): 96,15%.
Тогда как чувствительность в моем паттерне 87,27%, а селективность 100%.
© Сергеева Ирина 2009-2011