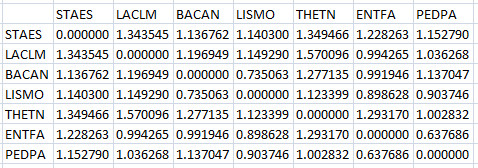
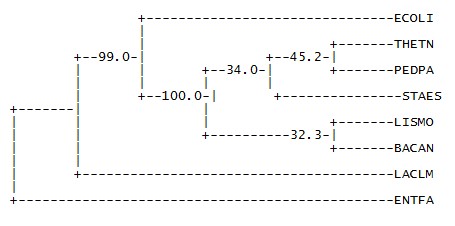Отобранные бактерии
| Название | Мнемоника |
| Bacillus anthracis | BACAN |
| Enterococcus faecalis | ENTFA |
| Listeria monocytogenes | LISMO |
| Lactococcus lactis | LACLM |
| Pediococcus pentosaceus | PEDPA |
| Staphylococcus epidermidis | STAES |
| Thermoanaerobacter tengcongensis | THETN |
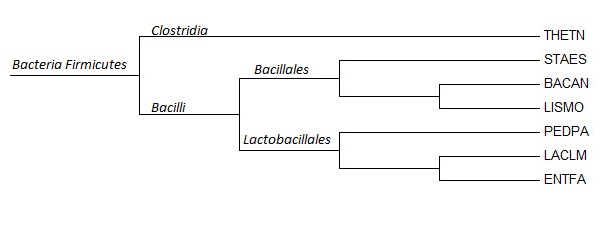
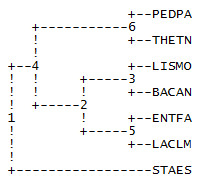
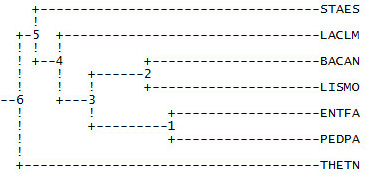
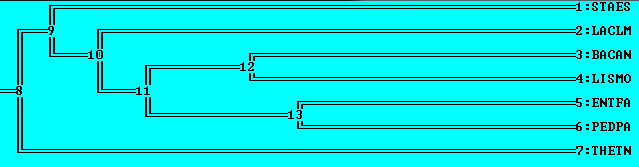
Скобочная формула дерева
(THETN,((STAES,(BACAN,LISMO)),(PEDPA,(LACLM,ENTFA))));
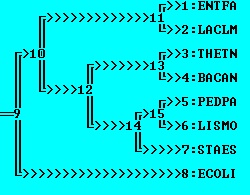
Изображение дерева

Ветви дерева
Дерево содержит четыре нетривиальные ветви:1) {BACAN, LISMO} против {THETN, STAES, PEDPA, LACLM, ENTFA};
2) {LACLM, ENTFA} против {THETN, STAES, BACAN, LISMO, PEDPA};
3) {STAES, BACAN, LISMO} против {THETN, PEDPA, LACLM, ENTFA};
4) {LACLM, ENTFA, PEDPA} против {THETN, STAES, BACAN, LISMO};