Изучение работы методов контроля температуры в GROMACS
Подготовление файла координат и файла топологии
С помощью make_ndx создал индекс-файл с группой, состоящей из одной молекулы этана. Для этого ввел "r 1" и нажал Enter. Создал файл .gro с одной молекулой с помощью editconf. Создал файл топологии, заменив цифру 38 на 1 в разделе molecules в фалй топологии et.top из предыдущего практикума.
Вычисления с помощью grompp и mdrun
Для вычислений были использованы пять методов контроля температуры:
- метод Берендсена для контроля температуры (be)
- метод "Velocity rescale" для контроля температуры (vr)
- метод Нуза-Хувера для контроля температуры (nh)
- метод Андерсена для контроля температуры (an)
- метод стохастической молекулярной динамики (sd)
Для каждого метода были проведены расчеты с помощью bash скрипта (две первые команды в цикле).
Анализ результатов
Для визуального анализа переконвертировал выходные файлы в pdb с помощью trjconv (третья команда в скрипте). Загрузил pdb файлы в PyMOL. Предварительные выводы:
- При использовании метода Берендсена молекула этана сначала ведет себя достаточно правдоподобно, хорошо прослеживаются колебания длины связи С-С, происходит вращение вокруг этой связи, но вскоре молекула начинает быстро крутиться, что не выглядит правдоподобно.
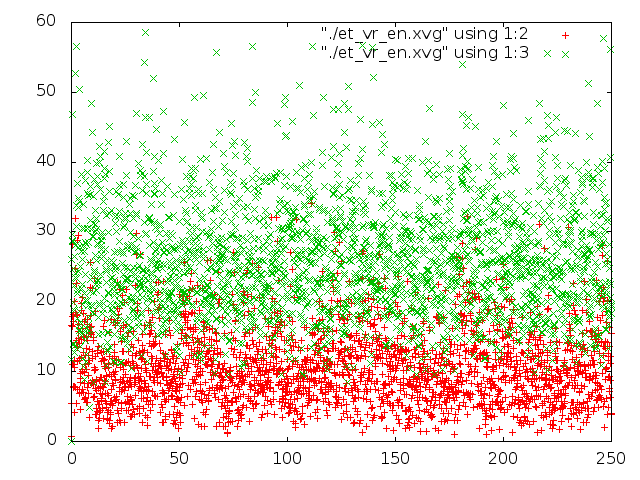
- Метод "Velocity rescale" ведет себя похожим образом, правда молекула крутится не так быстро.
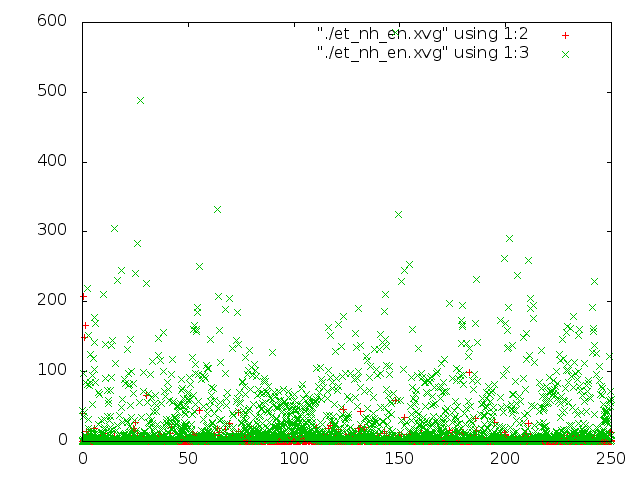
- При использовании метода Нуза-Хувера практически не происходит изменение длины связи C-C, но вращение вокруг нее очень реалистичное, иногда наблюдаются заслоненные конформации, но чаще заторможенные, как и должно быть.
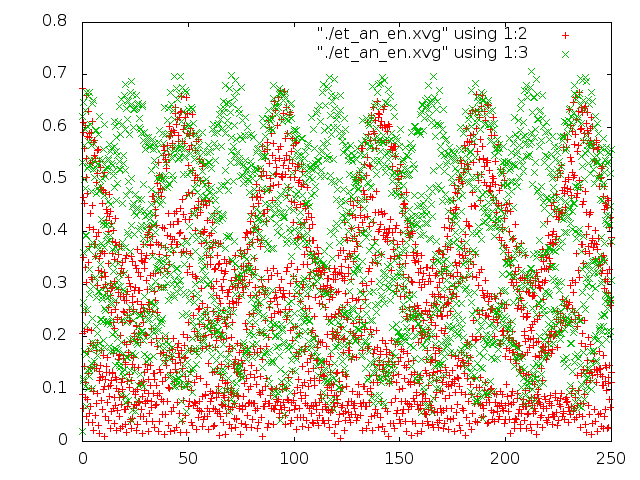
- Метод Андерсена меняет только длину связи, почти никакого вращения.
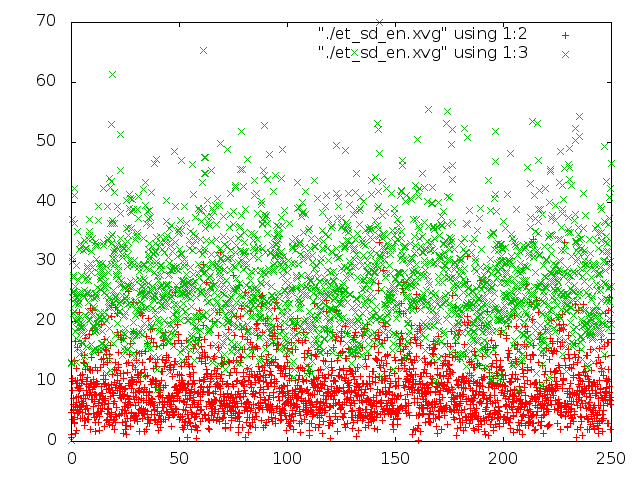
- После метода стохастической молекулярной динамики в поведении молекулы и правда появилось что-то стохастическое: каждое следующее положени молекулы сильно отличалось от предыдущего, проследить изменение длин связей и углов сложно.
Было проведено сравнение потенциальной энергии связи и кинетической энергии каждой из 5 систем с помощью g_energy. Потом были построены графики энергий с помощью gnuplot (четвертая и пятая команда в скрипте).
На графиках красным - потенциальная энергия связей, зеленым - кинетическая энергия.
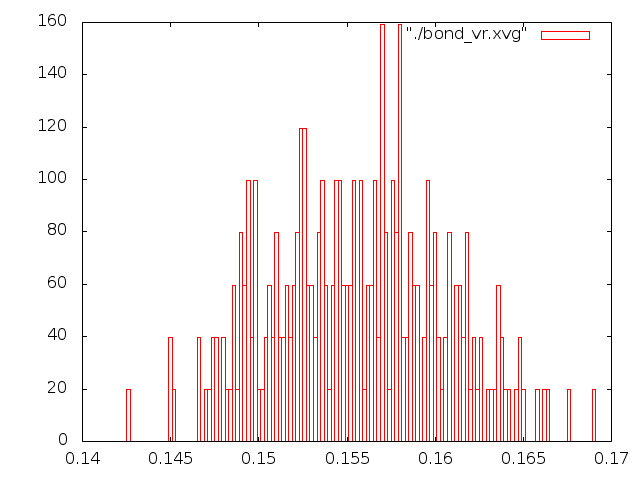
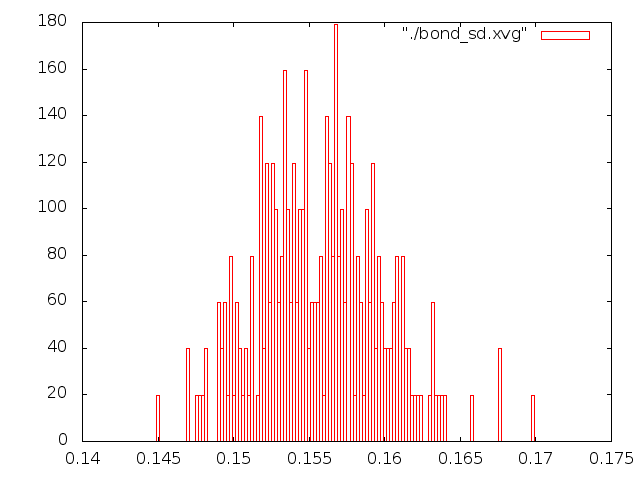
Исследовал распределение длины связи С-С за время моделирования с помощью g_bond, построил графики в gnuplot (две последние команды в скрипте). В идеальном случае это распределение должно быть похоже на распределение Максвелла-Больцмана.
Лучше всего распределением Больцмана описываюся методы "Velocity rescale" и стохастической молекулярной динамики, распределения для методов Нуза-Хувера и Берендсена на мой взгляд имеют слишком острый пик, т.е. длина связи меняется слабо, метод Андерсена не описывается распределением Больцмана вообще. Таким образом лучшим методом можно назвать "Velocity rescale", потому что поведение молекулы при контроле температуры методом стохастической молекулярной динамики показалось странным и, на первый взгляд, не поддающимся описанию.

2011
©
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}