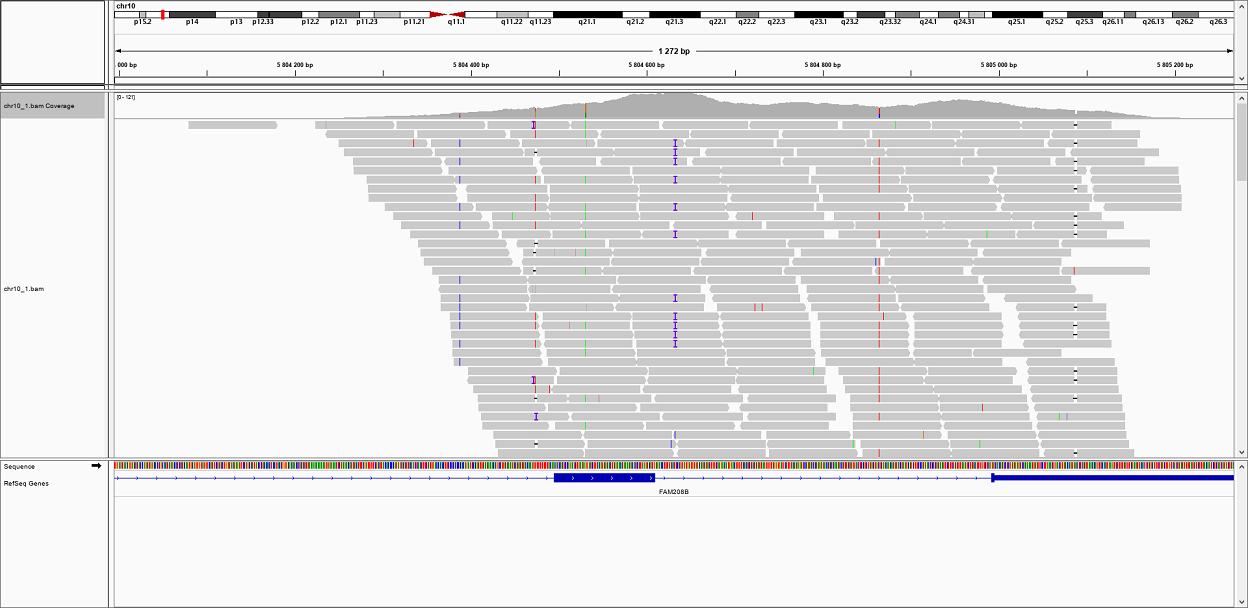
Изображение вставки (феолетовые I):
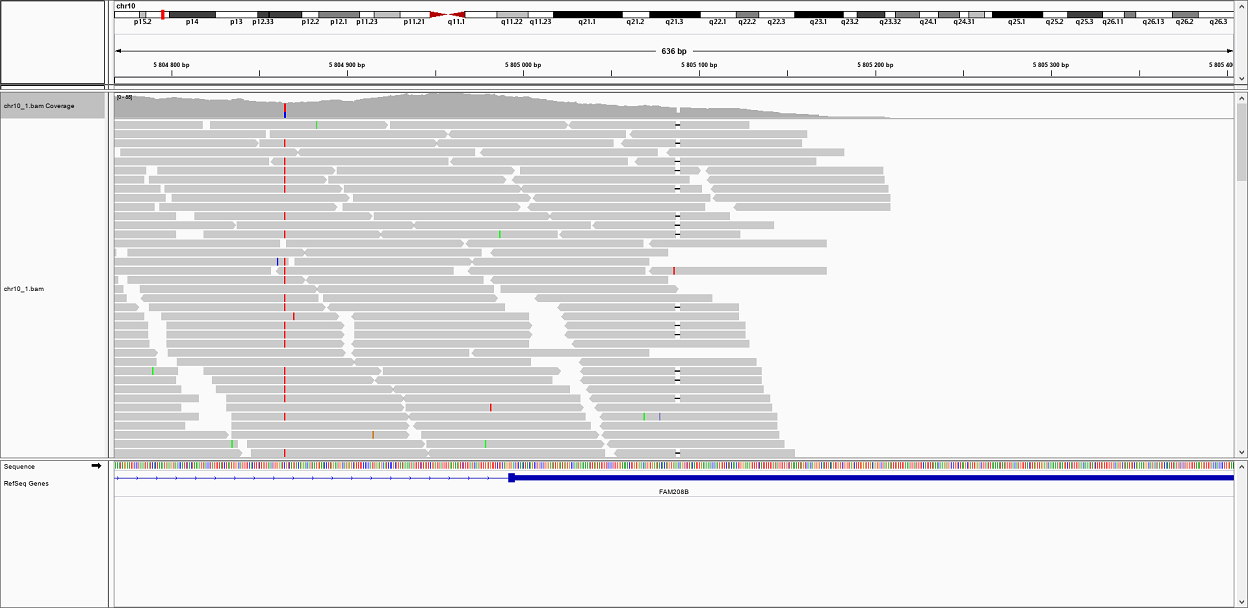
Изображение делеции (прочерки):
В этом практикуме мне досталась 10 хромосома. Для начала, последовательность была проиндексирована с помощью команды "hisat2-build chr10.fasta chr10".
Затем провели анализ данных одноконцевых прочтений 10 хромосомы с помощью программы fastqc (все команды в таблице 1). После этого с помощью программы trimmomatic с конца каждого чтения были отрезаны нуклеотиды с качеством ниже 20 и оставлены только чтения длиной не меньше 50 нуклеотидов. Затем был проведен их повторный анализ.
В результате, из 10666 последовательностей после использования trimmomatic осталось 10526. После использования этой программы улучшилось качество нуклеотидов с позициями 70 и выше, т.е. её использование оправдано.
| Команда | Результат |
| fastqc chr10.fastq | Zip-файл и файл html с анализом исходных чтений |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr10.fastq short2_chr10.fastq TRAILING:20 MINLEN:50 | Чтения с обрезанными с концов нуклеотидами, у которых качество больше 20 и длина более 50 нуклеотидов в файле short2_chr10.fastq |
| fastqc short2_chr10.fastq | Zip-файл и файл html с анализом чтений после использования trimmomatic |
Картинки из результатов fastqc с оценкой качества чтений нуклеотидов до и после использования trimmomatic:
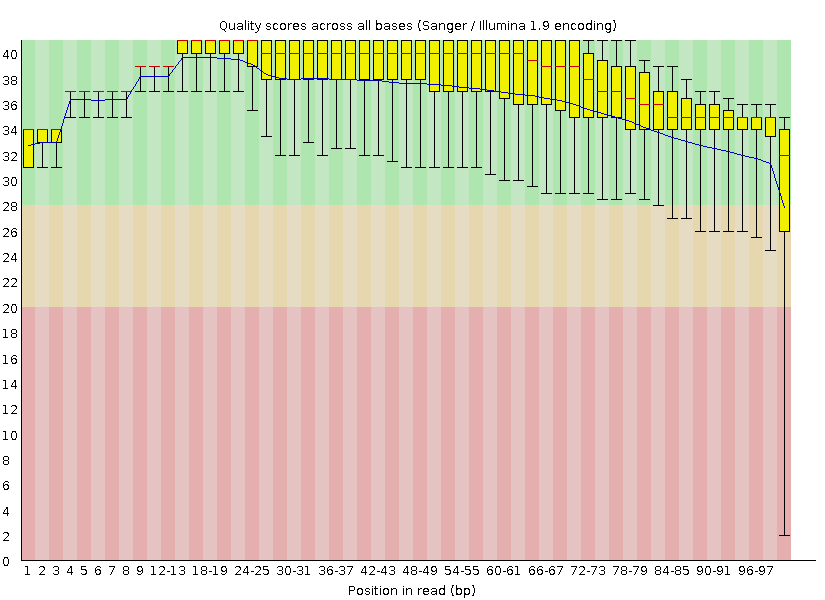
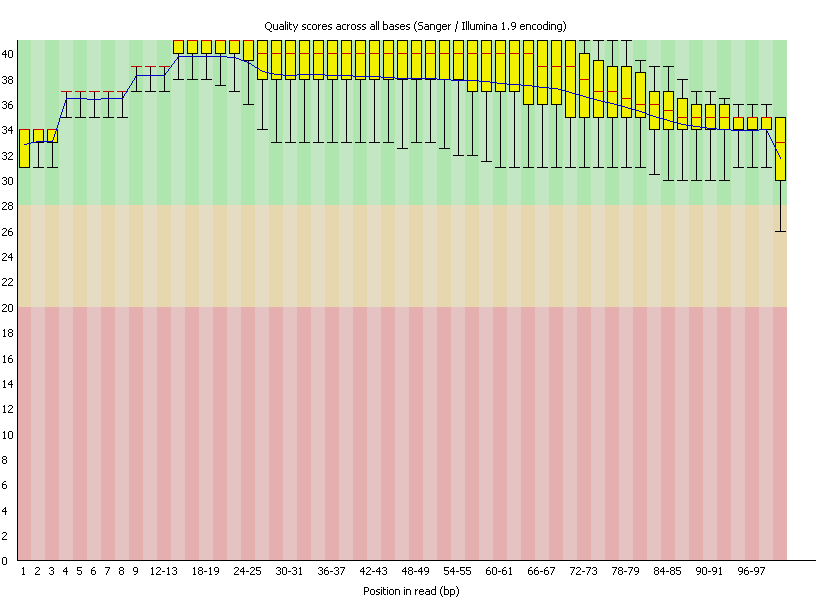
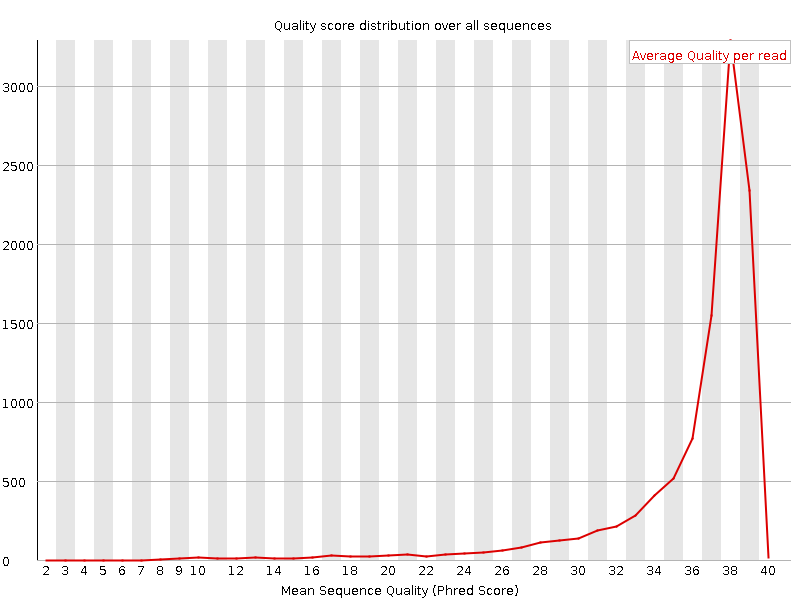
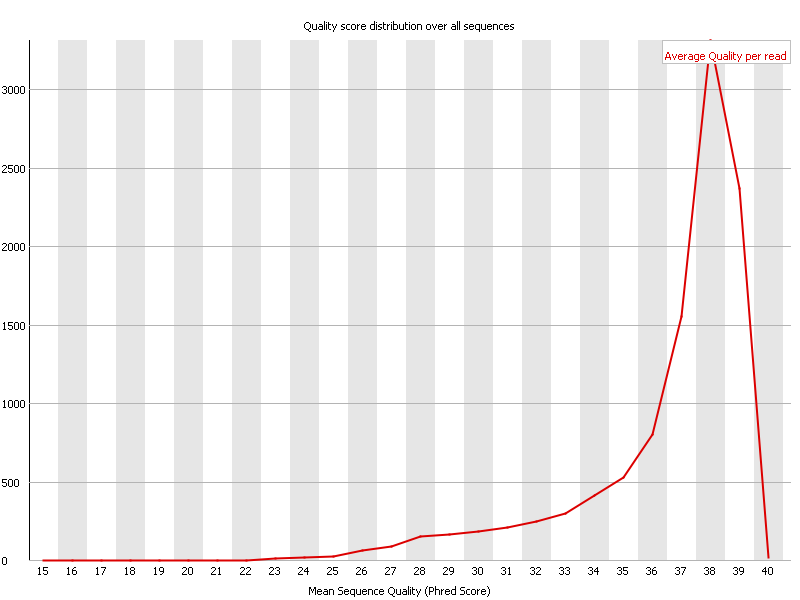
Картинки из результатов fastqc c оценкой качества чтений целых последовательностей до и после использования trimmomatic:
Для картирования чтений была использованна команда "hisat2 --no-spliced-alignment --no-softclip -x /nfs/srv/databases/ngs/ivanmike/chr10 -U /nfs/srv/databases/ngs/ivanmike/short2_chr10.fastq -S chr10_1.sam". Из полученного файла был создан бинарный с помощью команды "samtools view -b chr10_1.sam -o chr10.bam". Затем была проведена сортировка выравнивания чтений с референсом по координате в референсе начала чтения (команда "samtools sort chr10.bam chr10_1"). После этого полученный .bam файл был проиндексирован командой "samtools index chr10_1.bam". С помощью команды samtools flagstat оказалось, что 98.78% ридов из 10526 были картированы и 128 не были. Можно говорить о высоком качестве чтения ридов.
Для создания файла с полиморфизмами в формате .bcf была использована команда "samtools mpileup -uf chr10.fasta -g -o chr10_poly.bfc chr10_1.bam". Затем был создан файл со списком отличий между референсом и чтениями в формате .vcf (команда "bcftools call -cv chr10_poly.bfc -o chr10_poly.vcf"). С помощью этого файла была создана таблица 2 с 3 различными полиморфизмами.
| Координата | Тип | Референс | Чтение | Глубина покрытия | Качество чтений | |
| 1 | 5727202 | Замена | C | T | 5 | 43,0073 |
| 2 | 5804633 | Вставка | TTC | TTCTC | 119 | 217,468 |
| 3 | 5805087 | Делеция | AACA | AA | 37 | 217,468 |
Среднее значение качества чтений - 97,47, медиана - 69. Среднее значение глубины покрытия - 21,62, медиана - 8. Как видно около 50% чтений имеют качество меньше среднего. Медиана глубины покрытия показывает, что около 50% чтений были наложены на геном 8 раз.
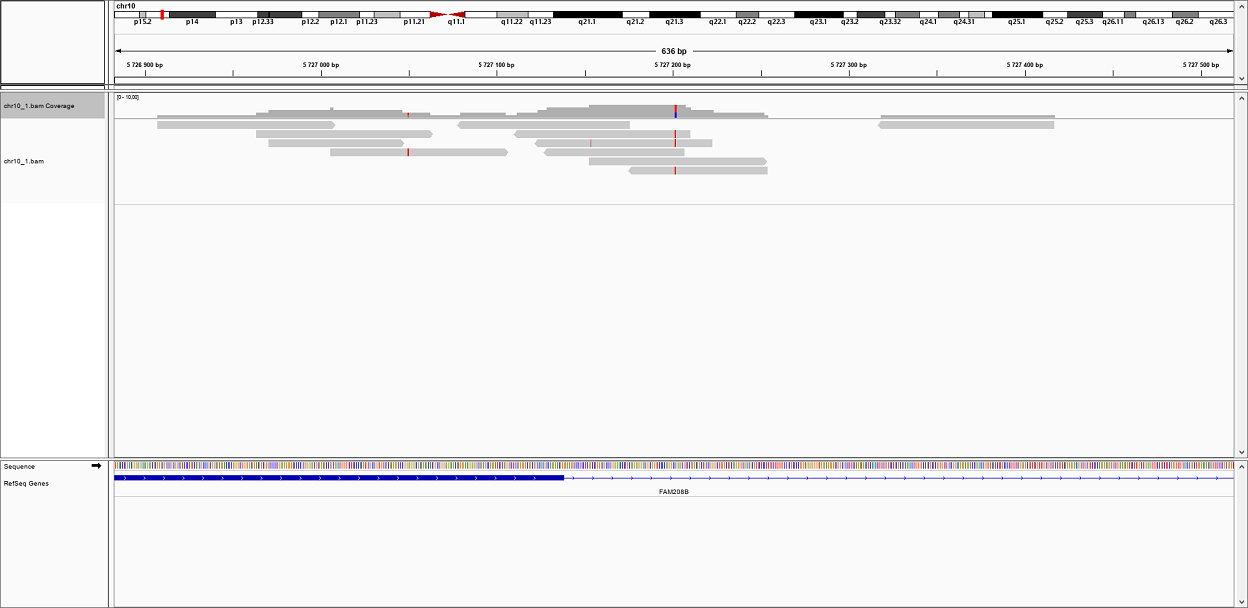
С помощью программы IGV были получены изображения данных полиморфизмов. Изображение замены (красные полоски):
Изображение вставки (феолетовые I):
Изображение делеции (прочерки):
Для начала был получен файл, скоторым может работать программа annovar с помощью команды "convert2annovar.pl -format vcf4 chr10_poly.vcf -o chr10_poly.avinput". Она показала 57 snp из которых 35 являются транзициями, 22 трансверсии и еще 9 инделей. Затем производилась аннотация по разным базам:
Можно сделать вывод, что клиническая аннотация snp недостаточная. Имеется информация только в базе Gwas о 3 snp, связанных с остеосаркомой, ревматоидным артритом и витилиго.
Назад