Для начала были проанализированны чтения одной из биологических реплик 10 хромосомы.
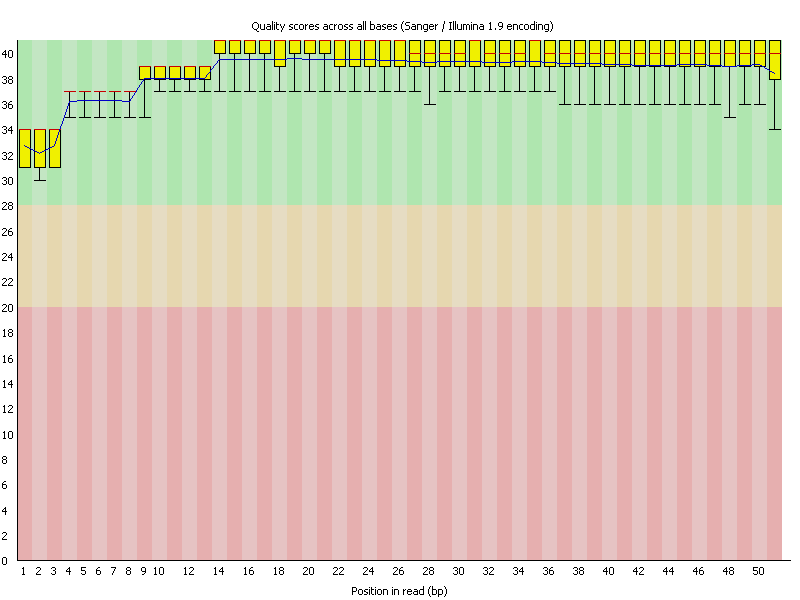
Как видно чтения имеют хорошее качество и не нуждаются в триммировании. Затем было проведено картирование чтений, в результате которого 98,67% из 15462 ридов оказались картированы, а 205 нет. После этого полученный файл в формате .sam был переведен в бинарный файл формата .bam, была проведена сортировка ыравнивания чтений с референсом по координате в референсе начала чтения, и полученный отсортированный был проиндексирован.
Для того чтобы узнать на какие гены легли прочтения была использована программа htseq-count. После выделения всех строчек с генами, у которых было 0 ридов был получен файл с содержимым:
ENSG00000165732.8 14643
ENSG00000266122.1 3
__no_feature 611
__not_aligned 205
Из него видно, что большая часть ридов связана с одним геном, и лишь 3 из них легли на другой. Для 611 чтений не нашлось границ генов, и 205 которые вообще не были картированы. Границ генов могло не найтись, если риды, например, содержали промоторную область гена. Ген ENSG00000165732.8 кодирует DEAD box белок, имеющий функцию РНК-хеликазы. Ген ENSG00000266122.1 в свою очередь является "отозванным" псевдогеном, который перекрывался с геном DEAD box белка.
Таблица 1 с использованными командами представлена ниже:
| Команда | Результат |
| fastqc chr10.1.fastq | Zip-файл и файл html с анализом исходных чтений |
| hisat2 --no-softclip -x /nfs/srv/databases/ngs/ivanmike/chr10 -U /nfs/srv/databases/ngs/ivanmike/chr10.1.fastq -S chr10.1.sam | Картирование чтений и вывод наложений на геном в файл chr10.1.sam. Опция --no-spliced-alignment оказалась не нужна, т.к. могли прочитаться экзоны, которые в геноме были разделены интроном и поэтому чтение может лечь не полностью (с разрывом на месте интрона). |
| samtools view -b chr10.1.sam -o chr10.1.bam | Перевод файла из формата .sam в бинарный формат .bam |
| samtools sort chr10.1.bam chr10.1_1 | сортировка выравнивания чтений с референсом по координате в референсе начала чтения |
| samtools index chr10.1_1.bam | Индексирование ридов |
| htseq-count -f bam -s no chr10.1_1.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseq | Подсчет ридов для каждого из генов. Опция -f - формат входного файла, -s - по какой из цепей выравнены чтения (значение no означает неопределенность направления), -i - атрибут, использующийся как feature ID (по-умолчанию нужный), -m - режим для неожнозначных выравниваний (по-умолчанию нужный) |
| grep -wv 0 htseq > htseq.txt | Выделение в отдельный файл всех строчек с генами, у которых было 0 ридов был получен файл с содержимым |