Практикум 9.
Задание 1. Сравнить состав систем рестрикции-модификации, закодированных в двух штаммах одного вида
Задача - сравнить предполагаемые (по избеганию сайтов) наборы систем Р-М в полном геноме бактерии из NCBI и наборе контигов того же вида из метагенома кишечника человека.
Этап 1. Нахождение избегаемых сайтов рестрикции в геноме выданной бактерии
Выданная бактерия - Bifidobacterium dentium (GenBank CP001750.1)
Для подсчета ожидаемого количества и контраста всех сайтов из списка в геноме бактерии, я воспользовалась веб-сервисом. Использовался метод Карлина. Результат представлен в файле.
Далее я перевела результаты в excel-таблицу и нашла все сайты, для которых контраст меньше чем 0.78 (порог, чтобы отличие от 1 можно было считать значительным).
В результате оказалось, что только для одной системы модификации-рестрикции порог меньше 0.78 - и равен 0,472. Соответственно, можно заключить, что эта система имеется у данной бактерии. Данные о ней записаны в файле файле.
Этап 2. Нахождение избегаемых сайтов рестрикции в наборе контигов из метагенома кишечника человека
Те же действия были проделаны для контигов генома этой бактерии из метагенома кишечника человека.
Результат работы сервиса представлен в файле.
Оказалось, что в бактерии из кишечника тоже присутствует только одна система МР, причем та же самая. Однако значение Karlin's ratio несколько больше и равно 0,569. Информация о данной системе МР находится в файле.
Обсуждение
В обеих бактериях обнаружилась только одна система модификации-рестрикции, причем одна и та же. Как я выяснила из соответствующей записи базы данных Nucleotide, бактерия, чей геном был секвенирован (из пункта 1) обитала в ротовой полости человека, тогда как бактерия из пункта 2 - в кишечнике человека. К сожалению, различий в числе и качестве систем модификации-рестрикции в данном случае не было обнаружено. Однако по одной этой паре бактерий никакого заключения относительно связи систем МР и местообитания сделать нельзя. Вполне возможно, что при проведении исследования на большой выборке выяснится, что такая связь существует.
Задание 2. Найдите последовательности Шайн-Дальгарно в геноме бактерии или археи, данном вам в первом семестре.
Я работала с археей Picrophilus torridus DSM 9790.
Литературные данные
Я нашла ряд статей и страниц, относящихся к данной теме:
Общая информация: http://parts.igem.org/Help:Ribosome_Binding_Sites/Shine-Dalgarno_sequence
Литературных данных о SD в геноме моей бактерии не нашлось, и вообще данных по моей архее в этом плане очень немного, есть только одна статья о публикации секвенированного генома.
Подготовка данных
С GenBank я скачала fasta файл с хромосомой Picrophilus torridus и таблицу особенностей (features). С помощью двух скриптов, предоставленных А.В. Алексеевским, я получила короткие последовательности, в которых стоит искать SD. Первый скрипт получает из таблицы особенностей координаты CDS, а второй по списку выбранных координат начала и конца искомой последовательности и файлу с хромосомой получает эти последовательности.
Для создания позиционной матрицы весов (PWM) с помощью MEME я взяла 297 самых длинных CDS и получила для них последовательности от -15 до 1 нуклеотида (от начала CDS = старта трансляции), руководствуясь данными из литературы. Для поиска SD по всему геному я получила последовательности для всех CDS от -30 до 1 нуклеотида. Такой диапазон был выбран задним числом: вначале я поискала SD в последовательностях от -20 до 1 и получила для распределение количества находок в зависимости от позиции начала SD. Даже среди находок с хорошим p-value (<0.02) было значительное количество таких, которые начинались на -20 позиции от старта трансляции, поэтому я решила расширить диапазон поиска.
Запуск MEME
Я запускала MEME со следующими параметрами: длина мотива - от 4 до 10 н., поиск только по данной цепи (поскольку даны кодирующие последовательности), в последовательности ожидается от 0 до 1 появления мотива.

Вначале я проводила поиск до нахождения 3-х разных мотивов, чтобы сравнить e-value. Результаты:

Первый их этих мотивов - искомая последовательность Шайна-Дальгарно. Видно, что e-value первого найденного мотива намного меньше, чем e-value второго и третьего.
Затем я запустила MEME для поиска одного мотива и получила позиционную матрицу весов для мотива SD:
Motif 1 position-specific probability matrix -------------------------------------------------------------------------------- letter-probability matrix: alength= 4 w= 6 nsites= 32 E= 1.7e-021 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 1.000000 0.000000 0.281250 0.000000 0.031250 0.687500 0.000000 0.000000 1.000000 0.000000 0.500000 0.031250 0.468750 0.000000 0.000000 0.125000 0.000000 0.875000
Запуск FIMO
Со страницей с html-выдачей MEME я перешла на страницу FIMO и запустила поиск мотива по файлу с последовательностями (от -30 до 1 нуклеотида, считая от старта трансляции) для всех генов моей археи - всего 1547 последовательности. Поиск проводился только по данной цепи.
Результаты: 173 находок с p-value <0.001, 580 с с p-value <0.01, 1471 с p-value <0.05, около 2700 с p-value <0.01. Поскольку у Picrophilus torridus всего 1547 генов, кодирующих белки, в идеальном случае SD должно было найтись примерно столько же. Однако последовательность короткая, поэтому в результатах поиска ожидатся много случайных находок. А также какое-то количество реально существующих последовательностей не будет найдено. В итоге я решила остановиться на пороге p-value <0.05 и 1471 находках. (Еще раз оговорюсь, что в этом списке не все SD из генома (но хотя бы большая часть) и есть случайные последовательности).
Для этих 1471 находок я построила LOGO:
![]()
Также для этих 1471 находок я построила распределение по началу относительно старта трансляции:
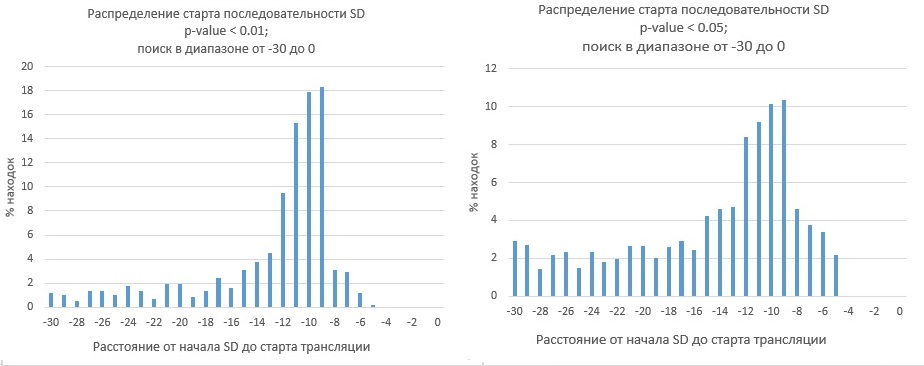
Видно, что хорошие находки (с p-value <0.01) в основном имеют начало в области от -9 до -12 (и заканчиваются, соотвественно, в области от -4 до -6). Это хорошо соответствует литературным данным (см. ниже). При увеличении p-value до 0.05 пик распределения по-прежнему находится там же, однако процент находок с началом раньше -9 и позже -12 возрастает. Возможно, это связано с накоплением случайных находок. (Однако напомню, что находок с p-value <0.05 - 1471, и это даже несколько меньше количества белок-кодирующих генов, поэтому выкидывать еще часть находок я не стала).
Ниже на рис.5 приводится изображение из статьи, посвященной поиску оптимального расстояния между SD и местом начала трансляции у E.coli:
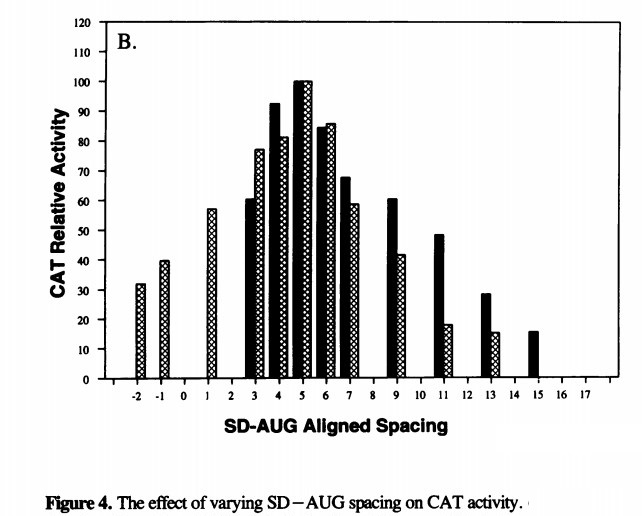
Если привести формат оси абсцисс к одному виду, видно, что распределение SD из этой статьи о E.coli очень похоже на полученное мной для P.torridus.
Изображение из другой статьи также показывает подобное распределение для E.coli и археи Pyrococcus abyssi:
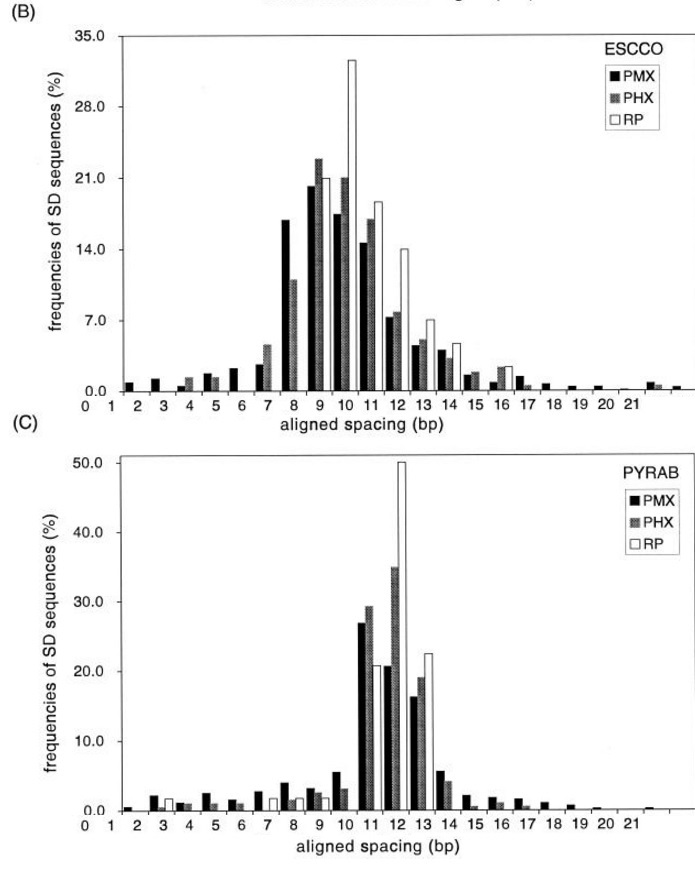
Замечу, что исходно на оси абсцисс было отложено расстояние от середины последовательности SD (GGAGG) до старт-кодона. Я сдвинула ось так, чтобы на ней было отложено расстояние от начала SD до старт-кодона (и обозначения совпадали с моими).
Несмотря на то, что P.torridus филогенетически ближе к архее Pyrococcus abyssi, чем к бактерии E.coli, ее распределение SD больше похоже на распределение для E.coli.
Надо заметить, что на диаграммах с рисунка 4 видно, что для рано начинающихся SD вероятность начала в данной позиции выше, если нуклеотид является первым в триплете в той же рамке считывания, что и CDS. Для SD, начинающихся с -18 позиции и позже, такой зависимости нет. Может быть, это связано с особенностями регуляции трансляции с помощью альтернативных сайтов посадки рибосомы.