| Главная | На страницу выбора семестров | Второй семестр |
Практикум 10
-
Задание 1
В этом задании я построил выравнивание 6 белков из семейства HSP70, по 2 из архей (DNAK_HALLT, DNAK_HALMA), бактерий (DNAK_MYCTO, DNAK_STRCO), эукариот (HSP7C_RAT, HSP7C_HUMAN). Я использовал программу JalView, выравнивание TcoffeeWS, раскраску ClusterX, установил порог идентичности 100%. С помощью программы infoalign из пакета EMBOSS я рассчитал количество консерватиыных а.к.о., функционально консервативных позиций, позиций, консервативных на 70%. Полное выравнивание на рисунке 1, ссылка на выравнивание. На выравнивании я разметил несколько позиций, консервативных на 80% и более (C), функционально консервативных (F), позиций с гэпами (G). Так, в позициях 6, 7, 17, 28 функциональная консервативность обуславливается наличием гидрофобных а.к.о. аланина, валина, изолейцина и лейцина. В позиции 162 присутствуют функционально идентичные остатки аспарагиновой и глутаминовой кислот, имеющих положительный заряд при физиологических pH. В таблице 1 представленые результаты расчета параметров выравнивания с помощью команды infoalign пакета EMBOSS. Всего абсолютно консервативных позиций в выравнивании 218, абсолютно функционально консервативных - 337. Стоит отметить, что длина гэпов, их расположение, а также параметры неполной консервативности для пары белков, взятых из одной систематической группы, схожи или идентичны, например, как для человеческого и крысиного белка.
Рис.1 Полное выравнивание.
Талбица 1. Абсолютно консервативные позиции.Имя Длина последовательности Длина выравнивания Длина гэпов Гэпы, % Абсолютно консервативные позиции АКП, % Функционально консервативные позиции ФКП, % Позиции, консервативные на 70% и более ПК70, % DNAK_HALLT_1-644 644 688 44 6,40 218 31,69 337 48,98 357 51,89 DNAK_HALMA_1-635 635 688 53 7,70 218 31,69 337 48,98 358 52,03 DNAK_MYCTO_1-625 625 688 63 9,16 218 31,69 337 48,98 365 53,05 DNAK_STRCO_1-618 618 688 70 10,17 218 31,69 337 48,98 357 51,89 HSP7C_RAT_1-646 646 688 42 6,10 218 31,69 337 48,98 330 47,97 HSP7C_HUMAN_1-646 646 688 42 6,10 218 31,69 337 48,98 330 47,97
-
Задание 2.
Для моделирования возникновения мутаций в белке я выбрал гистон H4 человека (UniProtID - H4_HUMAN), длина которого составляет 103 а.к.о. Текст скрипта, добавляющего 7 случайных мутаций в каждом поколении. Для построения выравнивания использовал программу JalView, алгоритм TcoffeeWS, раскраску ClusterX, установил порог идентичности 100%. Неисправленное выравнивание и исправленное выравнивание. В таблице 2 указаны первые 10 возникших мутаций, которые я описал после исправления.
Рис.2 Неисправленное выравнивание.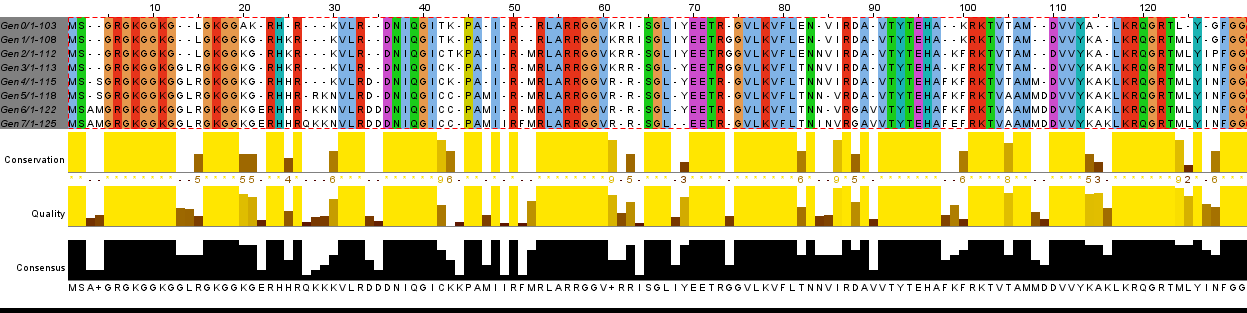
Рис.3 Исправленное выравнивание.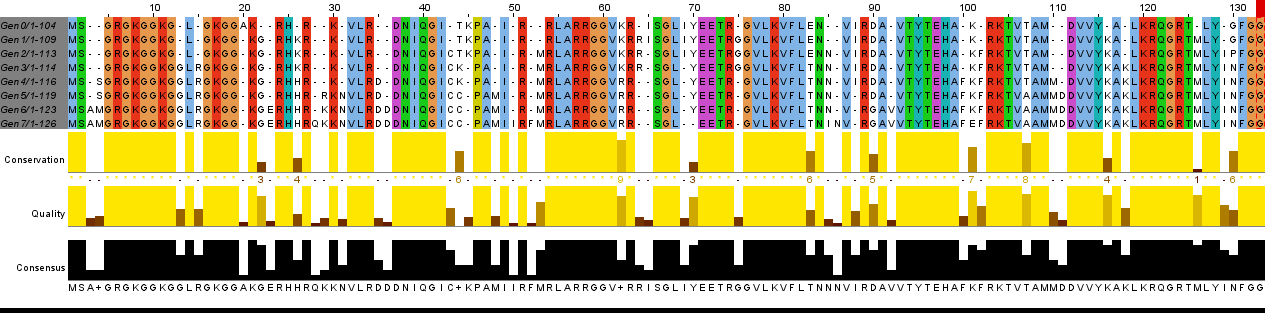
Таблица 2. Первые 10 мутаций.Было, АКО(Поз.) Стало, АКО(Поз.) Поколение мутации SG(2,3) SSG(2,3,4) g3-g4 S(3) M(3) g5-g5 SSG(2,3,4) SAMG(2,3,4,5) g5-g6 GL(10,11) GGL(10,11,12) g2-g3 LG(11,12) LRG(12,13,14) g2-g3 GAK(15,16,17) GK(15,16) g0-g1 KR(17,18) KGR(16,17,18) g0-g1 HR(19,20) HKR(19,20,21) g0-g1 GR(20,21) GER(21,22,23) g5-g6 HKR(21,22,23) HHR(22,23,24) g3-g4
Я перенес лейцины из позиции 14 выравнивания в позицию 13, чтобы была идентичность. В позицию 20 вставил гэпы 1-7 поколениям, чтобы получилась полностью консервативная колонка лизинов. Перенес лизины из 30 позиции в 29. -
Задание 3.
Для моделирования мутаций в кодирующей последовательности я использовал последовательность белка фотосистемы 2 Клевера лугового (GenBank: KX538828.1). Последовательность имеет длину 111 пар нуклеотидов, кодирует белок из 36 а.к.о. Для получения мутаций был написан скрипт, с помощью программы transeq пакета EMBOSS получена последовательность белка после введения мутаций в кодирующую последовательность. На рисунке 4 показано выравнивание алгоритмом TcoffeeWS, на рисунке 5 выравнивание ProbconsWS, в обоих случаях раскраска - Clustalx. Второе выравнивание показывает стоп-кодоны, которые появились в середине последовательности после внесения мутаций. Видно, что происходит сдвиг рамки считывания из-за однонуклеотидных вставок и делеций, а это приведет , скорее всего, к потери функции белка. В этом случае говорить о выявлении места мутаций не получится, так как сложно построить выравнивание, отражающие эти изменения. Для наглядности на рисунке 6 показано выравнивание (алгоритм TcoffeeWS раскраска Clustalx) последовательностей ДНК. Видно, что в последнем поколении помимо стоп-кодонов в последовательности первый кодон подвергся мутации и теперь кодирует аспарагин.
Рис.4 Полученное выравнивание, алгоритм TcoffeeWS.
Рис.5 Полученное выравнивание, алгоритм ProbconsWS.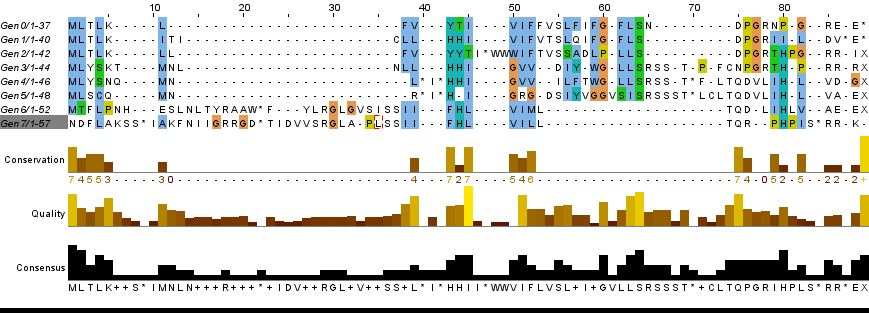
Рис.6 Выравнивание последовательности ДНК.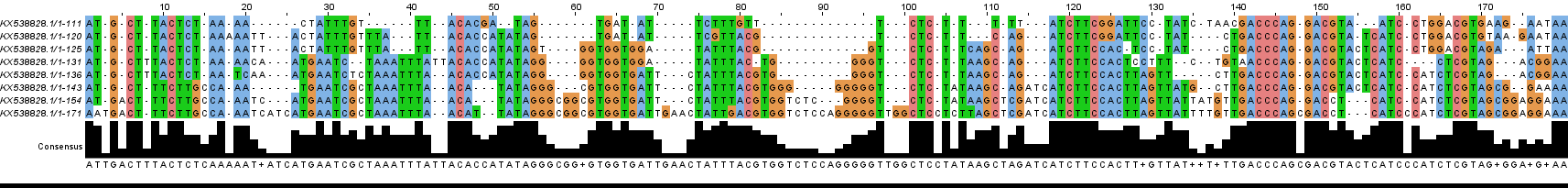
-
Задание 4.
Для белков есть проверка - сходство структур. Несомненно, есть схожие белковые структуры, образованные из гомологичных структур. Однако сходство белков может быть результатом конвергенции, как и в случае с макроскопическими структурами организмов на других, более высоких, уровнях жизненной организации (плавники рыб и вторичноводных млекопитающих).
Эволюционирует нуклеотидная последовательность генома. Нуклеотидная последовательность подвергается факторам мутагенеза и ошибкам при передаче наследственной информации от родительской клетки к дочерней (ошибки репарации, репликации и тд.). Эти же ошибки определяю изменение белков, их свойств и всего организма. Именно фенотип организма подвергается естественному отбору, мутации закрепляются, если определяемые ими изменения способствуют выживанию и размножению организма.
Только мутации в половых клетках наследуются. Организмы, способные к вегетативному и бесполому размножению, передают наследственную информацию с мутациями своим потомкам через соматические клетки. Например, при выращивании картофеля пользуются возмножностью вырастить растение из прошлогоднего клубня.
Страница находится на стадии разработки
© Poddyakov Ivan 2016-2018