Анализ транскриптомов.
- Задание 1. Освоение команд
В данном практикуме я работал с хромосомой 17 человека. Все команды выполнены в моей рабочей директории /nfs/srv/databases/ngs/ivanpodd .
Команда Результат cp ../Human/rnaseq_reads/chr17.1.fastq chr17_1.fastq Копирование файла в рабочую директорию fastqc chr17_1.fastq Анализ качества чтений java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr17_1.fastq trim17.fastq TRAILING:20 MINLEN:50 Очистка чтений: отрезаем позиции с качеством меньше 20 и убираем последовательсти с длиной меньше 50 fastqc trim17.fastq Анализ качества чтений после триммирования hisat2 -x index17 -U trim17.fastq -S trim17align.sam --no-softclip Картирование чтений на референс samtools view -b trim17align.sam -o trim17align.bam Конвертация выравненных чтений в бинартый файл samtools sort trim17align.bam trim17alignsorted Сортировка выравнивания samtools index trim17alignsorted.bam Индексация сортированного бинарного файла htseq-count -f bam -s no -i gene_id -m union trim17alignsorted.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf -o count1.sam > count Подсчет чтений grep -wv 0 count > results.txt Собираем ненулевые строки из результата htseq Таблица 1.
- Задание 2. Результаты
Всего было 10407 ридов. После очистки чтений осталось 10355. Среднее качество прочтения для позиций до и после очистки приведены на Рисунке 1 и Рисунке 2 соответственно. После обрезки качество чуть-чуть улучшилось.
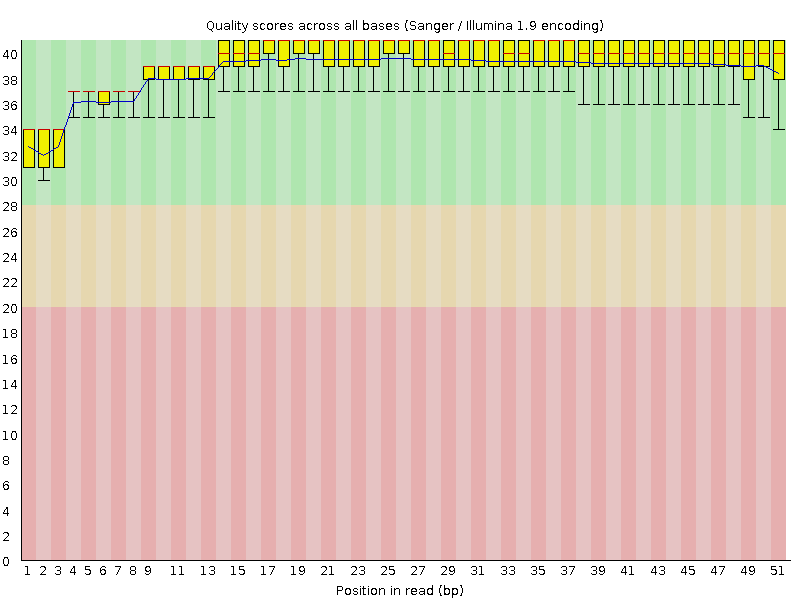
Рисунок 1.
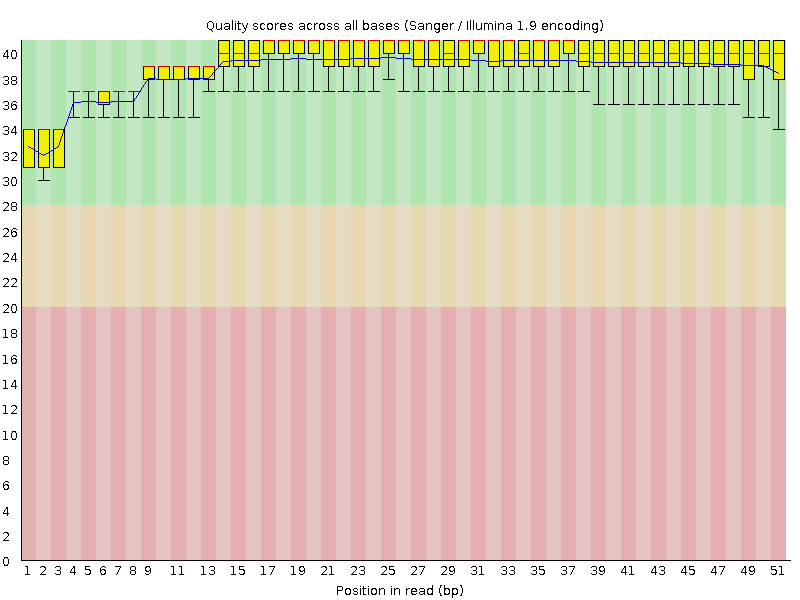
Рисунок 2.
После картирования 96,85% чтений выравнились на референс, из них 0% более одного раза. Похоже, что качество картирования хорошее. Я убрал параметр --no-spliced-alignment, так как мы работаем с транскриптомом, где как раз есть сплайсинг и где выравнивания могут разрываться.
Для запуска программы htseq-count использовались опции -f, -s, -i, -m. Опция -f указывает формат входного файла, опция -s говорит, должна ли программа учитывать цепь, -i - как программа назовет гены, -m - как программа будет реагировать на разное наложение рида на ген/гены. Выдача grep:
ENSG00000108424.5 8708 ENSG00000263766.1 1 __no_feature 414 __ambiguous 906 __not_aligned 326
8708 ридов укладываются на ген KPNB1 - karyopherin subunit beta 1. Кариоферины - белки, повлеченные в транспорт между цитоплазмой и ядром. 1 рид соответствует гену NPEPPS puromycin sensitive aminopeptidase - пиромицин-чувствительная аминопептидаза. Часть (414) ридов выравнились на участки, где никаких генов не показано. Так как я запускал htseq-count с опцией union, 906 ридов выравниваются частично или полностью на два гена. 326, как я понимаю, не выравнились никуда.