Секвенирование по Сэнгеру. Хроматограммы.
В данном практикуме было необходимо научится работать с результатами секвенирования по Сэнгеру. В ходе выполнения заданий я пользовался программой CodonCodeAligner.
- Задание 1. Чтение хроматограмм - возможные проблемы.
Прежде чем рассматривать проблемные нуклеотиды, нужно определить нечитаемые концы хроматограмм. В использованной программе есть встроенная функция, обрезающая "плохие" концы - clip ends. Результаты показаны на Рисунках 1, 2, где обрезанному началу/концу не сопоставлены буквы. Моя оценка отличается от программной, так как мне показалось, что накладывающиеся друг на друга пики (5' Reverse), слабо разделимые пики одного сигнала (5' Forward), пики разных сигналов в одной позиции, сильно ниже окружения по уровню, (3' Reverse) - ухудшают читаемость хромотограммы. Я отметил красным нуклеотиды, между которыми включительно я счел хромотограмму читаемой.
Таким образом, программа определяет первые 34 и 38 позиций в начале последовательности трудночитаемыми для F и R соответственно, 5 и 13 соответственно в конце. На мой взгляд, это согласуется с тем, что начало прочтения обычно содержит много шума, как остаточное количество краски, праймеров ПЦР и сиквенсовой реакций, а фрагменты длины около 380 нуклеотидов еще можно хорошо различить.
Полученная консенсусная последовательность, выравнивание, исходные F и R.
Приведу следующие примеры сложных позиций. В позиции 37 в последовательности R (Рисунок 3) показаны три разных пика G, A, C. Так как последовательность F недоступна на данном участке, думаю уместно поставить V - не T.
На Рисунке 4 показан участок, где шум на хроматограмме в последовательности F мешает определить нуклеотид. В последовательности R есть две пересекающиеся позиции, где также не установлен нуклеотид. В 111 позиции в обоих случаях есть пик C, несмотря на его слабость в R, считаю возможным определить ее как C. В позиции 115 контига в F есть все 4 сигнала, в R - очень слабые сигналы T и C. Думаю, можно обозначить как Y - пиримидиновое основание.
В позиции 235 на Рисунке 5 в последовательности Forward присутствует пик T и более слабый пик G. Мне кажется, что это не полиморфизм, а небольшой шум от соседних позиций G. Похожий случай - в позиции 382 (Рисунок 6), одноко тут нет данных о комплементарном прочтении, поэтому я думаю стоит поставить Y.
Сравнивая качество двух хроматограмм, "на глаз" F кажется более читаемой, так как в ней меньше участков с шумом, но на самом деле, в конце довольно продолжительный участок с перекрыванием пиков и разными сигналами в одной позиции, а программа считает F более качественной, чем R (315 против 311) - параметр quality.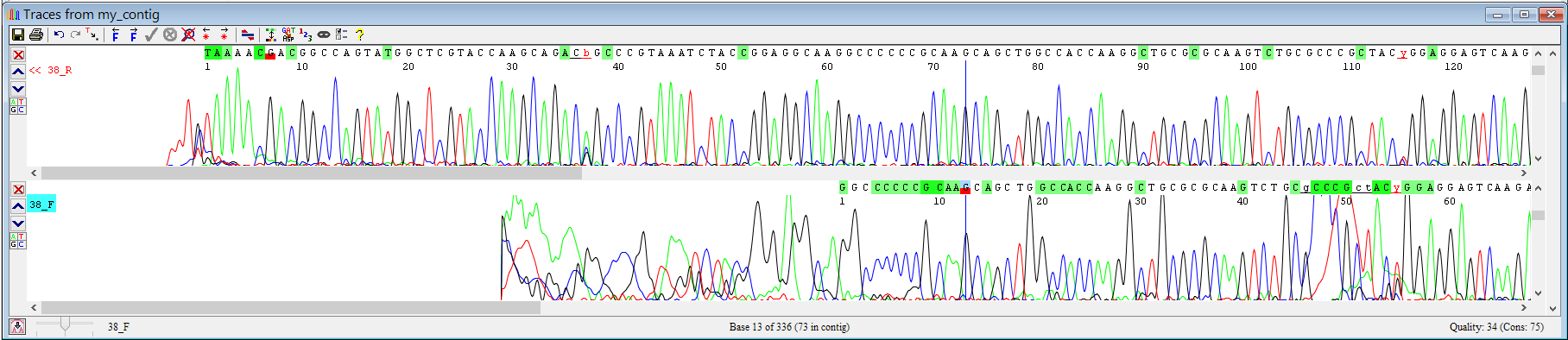
Рисунок 1. Начало контига.
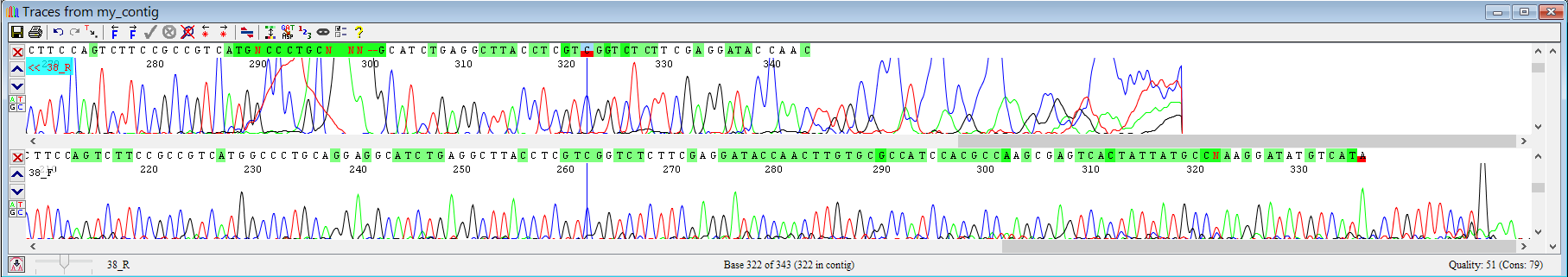
Рисунок 2. Конец контига.
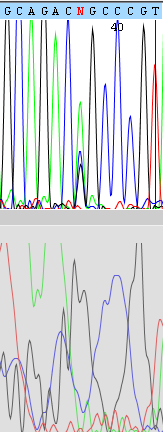
Рисунок 3.
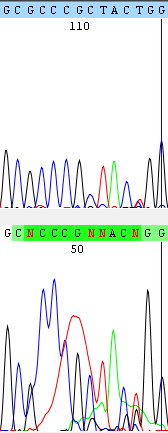
Рисунок 4.
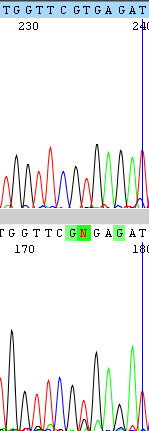
Рисунок 5.
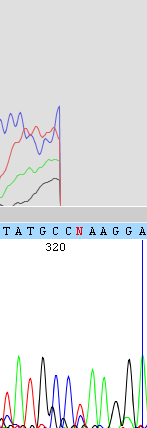
Рисунок 6.
- Задание 2. Пример нечитаемой хроматограммы.
Как пример нечитаемой хроматограммы я взял следующую пару F и R. Верхняя хроматограмма имеет качество 0, и программа не может определить основания и выровнять последовательности. Во второй хроматограмме, тем не менее, можно прочитать неокторые фрагменты. На выделенном участке снизу (Рисунок 7) видно чуть более низкие пики красного цвета поверх других. Возможно, это вызвано делецией нуклеотидов или ошибкой полимеразы. На верхней же хроматограмме нельзя выделить ни одного пика, так как они не только наложены друг на друга, но и появляются с нерегулярным шагом, то есть это может быть следствием наличия в пробе разных образцов ДНК, прошедших сиквенсовую реакцию с неспецифичными праймерами. Также предполагаю, что такая хроматограмма может быть следствием неисправности секвенатора.
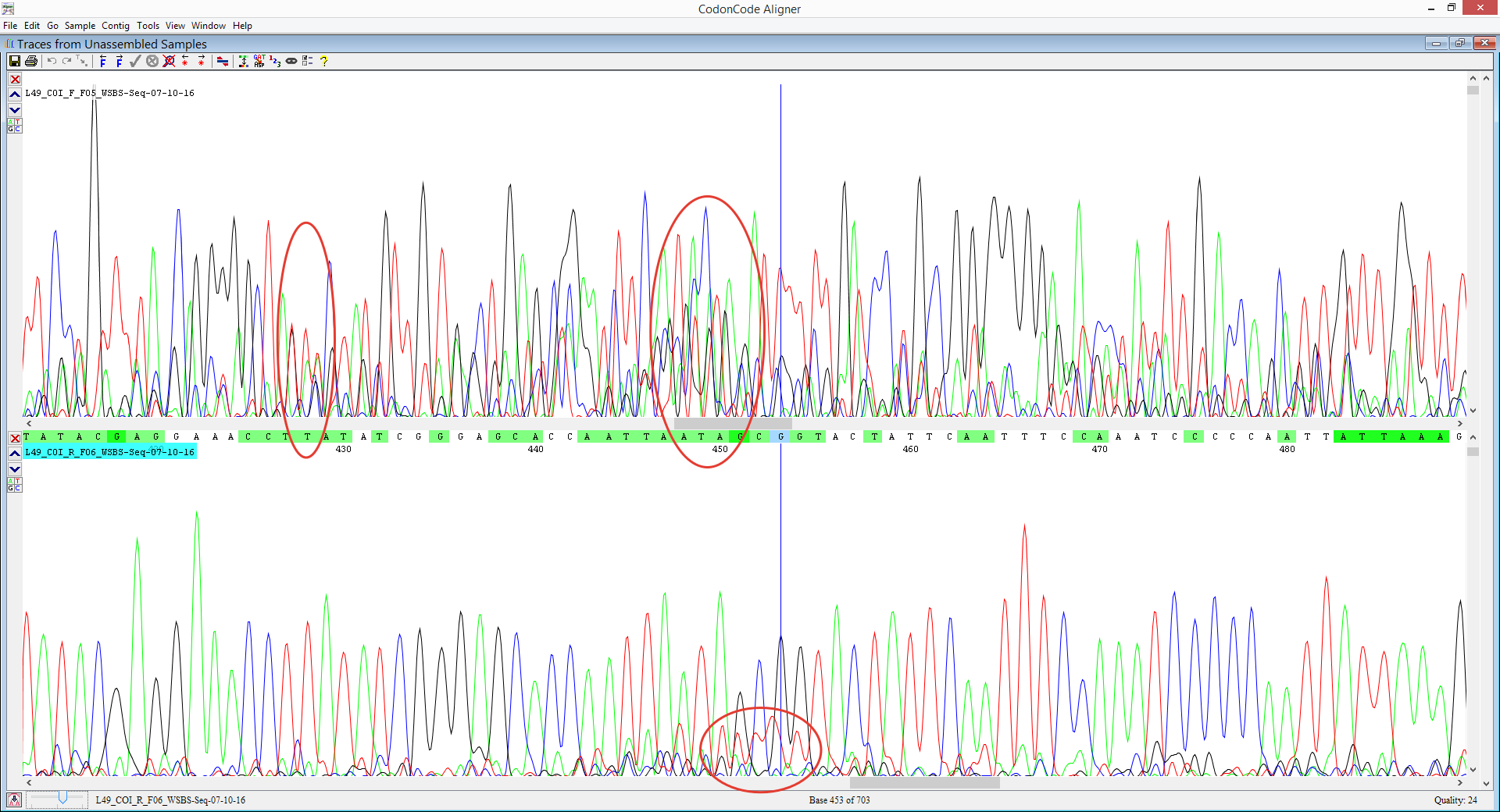
Рисунок 7. Фрагмент нечитаемой хроматограммы.