Узлы подписаны исходя из того, какие листья выходят из них;
Дерево построено в программе Paint.
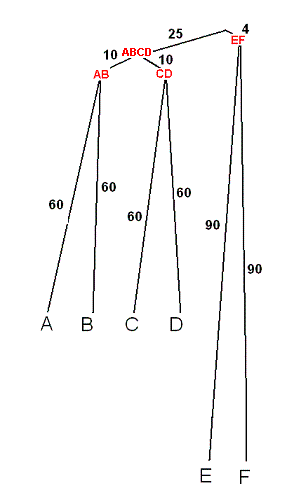
Модель судьбы моего гена описана в виде скобочной формулы
(((А:60,B:60):10,(C:60,D:60):10):25,(E:90,F:90):4);.
По ней было реконструировано дерево.
|
Длины ветвей даны как число мутаций на 100 нуклеотидов гена; Узлы подписаны исходя из того, какие листья выходят из них; Дерево построено в программе Paint. |
| A | B | C | D | E | F |
| . | . | * | * | * | * |
| * | * | . | . | * | * |
| . | . | . | . | * | * |
Считая дерево бескорневым, построена таблица разбиений. Cтолбцы соответствуют листьям дерева, а строки - ветвям дерева.
Разными знаками для внутренних ветвей обозначены листья находящиеся по разные стороны от этих ветвей. Поэтому нет
смысла описывать таким образом внешние ветви (т.к. они отделяют только один лист от всех остальных)
Получение искуственных мутантных последовательностей.
Следующим этапом было построение искуственных мутантных последовательностей, соответствующих
листьям и узлам дерева, считая, что в корне находится последовательность гена белка
Rho_ecoli.
Для того чтобы строить последовательности необходимо вычислить количество мутаций для каждого процесса (например, для получения
последовательности ABCD, соответствующей одноименному узлу дерева).
Количество мутаций для каждого процесса вычисляли по формуле: M=D*distance/100, где M-число мутаций, D-длина гена (для данного белка она равна 1260 нуклеотидов), distance-длина соответствующей ветви.
Написан скрипт script для программы
msbar пакета EMBOSS. Как видно из текста скрипта, исходной последовательностью является последовательнось гена RHO_ECOLI,
которая последовательно, в соответствии со строением дерева подвергается мутации. Причем в параметр -count задается количество
замен нуклеотидов посчитанное по формуле в соответствии с длиной той или иной ветви. Также в скрипте в каждой строке прописывается еще один параметр -
-point, который определяет тип точечной мутации, который следует сделать (так, кроме замены можно сделать вставку, делецию и тд.)
Чтобы сделать скрипт исполняемым используется команда :chmod +x script.txt
Запуск скрипта : ./script.txt
В конце концов получились 6 последовательностей,
соответствующих листьям дерева.
Построение деревьев.
Чтобы реконструировать дерево алгоритмом максимального правдоподобия, запустим программу fdnaml:
fdnaml all.fasta -ttratio 1 -auto, где параметром -ttratio задаем отношение транзиций и трансверсий, которое задает эволюционную модель.
Эту модель использует программа для подсчета вероятности получение исходного выравнивания мутированных последовательностей исходя из генерированного ею дерева.
В качестве выводного результата программа показывает дерево с наибольшей вероятностью получения на основе него и принятой эволюционной модели поданного на вход выравнивания.
Выходной файл fdnaml: all.fdnaml
Чтобы реконструировать дерево алгоритмами UPGMA или Neighbor-joining, сначала надо посчитать попарные
расстояния между последовательностями программой fdnadist:
fdnadist all.fasta -ttratio 1 -auto
Выходной файл all.fdnadist нужно подать на вход программе fneighbor
| Максимального правдоподобия | UPGMA |
+-------------B | | +-----------------------F | +------4 | | +--------------------E 1--3 | | +--------------D | +--2 | +-------------C | +---------------A |
+------------------------------A
+-2
! +------------------------------B
+-----------3
! ! +----------------------------C
+--4 +---1
! ! +----------------------------D
--5 !
! +--------------------------------------------E
!
+-----------------------------------------------F |
| Neighbor-joining | |
+--------------B ! ! +-------------C ! +-2 ! ! +---------------D 3-4 ! ! +--------------------E ! +------1 ! +------------------------F ! +---------------A |
Таблица сравнения.
| A | B | C | D | E | F | Исходное дерево | Максимальное правдоподобие | Neighbor-joining | UPGMA |
| . | . | * | * | * | * | + | + | + | + |
| * | * | . | . | * | * | + | + | + | + |
| . | . | . | . | * | * | + | + | + | + |