
<< BACK
Я построил 6-последовательное выравнивание по алгоритму ТcoffeeWSслучайно выбранных белков в семействе HSP70 из Eukaryota, Bacteria и Archea.Затем окрасил его по схеме ClustalX с условием Identity Threshold = 100% (представлено на рис. 1). Цветовая схема совершает окрашивание столбцов только со 100% идентичными аминокислотными остатками в каждой последовательности.
Рис 1. Полное выравнивание
На выравнивании я разметил несколько позиций,
консервативных на 80% и более (C), функционально консервативных (F), позиций с гэпами (G). Так, в позициях 6, 7, 17, 28 функциональная консервативность обуславливается наличием гидрофобных а.к.о. аланина, валина, изолейцина и лейцина. В позиции 162 присутствуют функционально идентичные остатки
аспарагиновой и глутаминовой кислот, имеющих положительный заряд при физиологических pH.
Также я определил основные свойства рассматриваего выравнивания с помощью пакета программ EMBOSS, для этого мною использовалось три различные
опции: -identity 100.0, -identity 70.0 и -plurality 100.0. Сначала продемонстрировано идентичные на 100% остатки, затем - на 70%, в итоге - на 100%
функционально идентичные остатки. Кроме этого, мною использовались функции для подсчета количества позиций, занимаемых "гэпами" (-gapcount) и
длины выравнивания (-alignlength). Полученные данные можно увидеть в таблице 1.
Uniprot AC |
Domain |
Sequence length |
Alignment length |
Amount of gap positions |
Percent of gap positions |
Amount of 100% conserved positions |
Percent of 100% conserved positions |
Amount of 70% conserved positions |
Percent of 70% conserved positions |
Amount of functionally conserved positions |
Percent of functionally conserved positions |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DNAK_HALLT_1-644 | Archaea | 644 | 688 | 44 | 6,4 | 218 | 31,69 | 337 | 48,98 | 357 | 51,89 |
| DNAK_HALMA_1-635 | Archaea | 635 | 688 | 53 | 7,7 | 218 | 31,69 | 337 | 48,98 | 358 | 52,03 |
| DNAK_MYCTO_1-625 | Bacteria | 625 | 688 | 63 | 9,16 | 218 | 31,69 | 337 | 48,98 | 365 | 53,05 |
| DNAK_STRCO_1-618 | Bacteria | 618 | 688 | 70 | 10,17 | 218 | 31,69 | 337 | 48,98 | 357 | 51,89 |
| HSP7C_RAT_1-646 | Eukaryota | 646 | 688 | 42 | 6,1 | 218 | 31,69 | 337 | 48,98 | 330 | 47,97 |
| HSP7C_HUMAN_1-646 | Eukaryota | 646 | 688 | 42 | 6,1 | 218 | 31,69 | 337 | 48,98 | 330 | 47,97 |
В таблице 1 представленые результаты расчета параметров выравнивания с помощью команды infoalign пакета EMBOSS. Всего абсолютно консервативных позиций в выравнивании 218, абсолютно функционально консервативных - 337. Стоит отметить, что длина гэпов, их расположение, а также параметры неполной консервативности для пары белков, взятых из одной систематической группы, схожи или идентичны, например, как для человеческого и крысиного белка.
Для моделирования возникновения мутаций в белке мной был выбран гистон H4 человека (UniProtID - H4_HUMAN), длина которого составляет 103 а.к.о. Текст скрипта bash, работающего на EMBOSS и добавляющего 7 случайных мутаций в каждом поколении. Для построения выравнивания я использовал программу JalView, алгоритм TcoffeeWS, раскраску ClusterX с порогом идентичности 100%. Неисправленное выравнивание по ссылке.
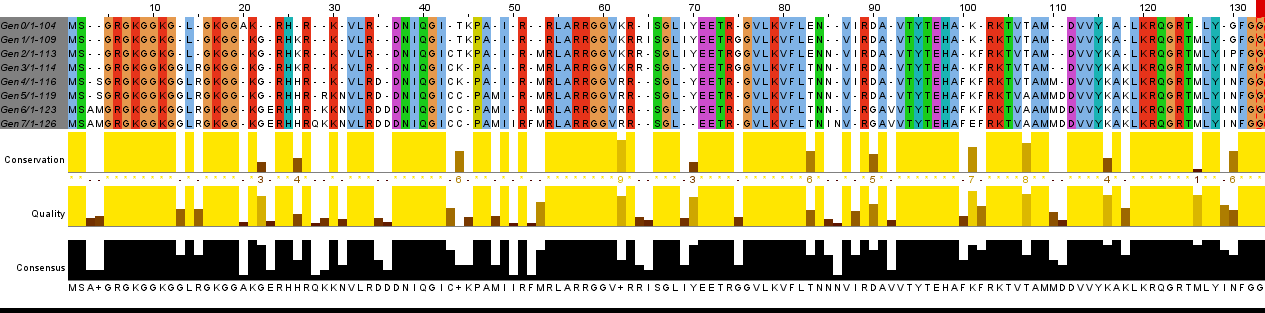
Рис 2. Исправленное выравнивание. Gen0- исходная последовательность, Gen1-Gen7 - видоизмененные потомки. TCoffeeWS с 100% идентичность.
Мутации были воспроизведены таким образом, что только семь точечных мутаций произошло в каждом из семи рассматриваемых пооколений. Информация о первых десяти позициях содержится в таблице 2.
| Было, а.к.о (поз.) | Стало, а.к.о (поз.) | Поколение мутации |
| SG(2,3) | SSG(2,3,4) | g3-g4 |
| S(3) | M(3) | g5-g5 |
| SSG(2,3,4) | SAMG(2,3,4,5) | g5-g6 |
| GL(10,11) | GGL(10,11,12) | g2-g3 |
| LG(11,12) | LRG(12,13,14) | g2-g3 |
| GAK(15,16,17) | GK(15,16) | g0-g1 |
| KR(17,18) | KGR(16,17,18) | g0-g1 |
| HR(19,20) | HKR(19,20,21) | g0-g1 |
| GR(20,21) | GER(21,22,23) | g5-g6 |
| HKR(21,22,23) | HHR(22,23,24) | g3-g4 |
Мной были перенесены лейцины из позиции 14 выравнивания в позицию 13, чтобы была идентичность. В позицию 20 вставил гэпы 1-7 поколениям, чтобы получилась полностью консервативная колонка лизинов. Перенес лизины из 30 позиции в 29. Подводя итог, можно утверждать, что выравнивание указывает лишь на возможный путь эволюции. Данный опыт показал, что эвистический алгоритм выравнивания не способен правильно восстановить даже простой пример процесса эволюции.
{kind=link}