Алгоритмы филогенетической реконструкции
В предыдущем практикуме мною были выбраны 15 организмов. Для них было построенно филогенетическое дерево, основанное на таксономии.
В рамках этого практикума разными способами были построены ещё 3 дерева, основанные на последовательности белка цитохрома b.
Подготовка данных
Первым делом были получены последовательности белков выбранных организмов:
seqret @cyb.list cyb.fasta
Тут файл cyb.list состоит из строк вида:
sw:cyb_brapc sw:cyb_pargo sw:cyb_loxaa ...
Далее белки последовательности (файл cyb.fasta) были выровнены с помощью программы muscle:
muscle -align cyb.fasta -output cyb-alignment.fasta
Далее я посмотрел выравнивания с помощью less, и они оказались нормальными.
Далее необходимо было перевести формат файла в .phy для построения дерева. Для этого был использован скрипт:
from Bio import AlignIO
in_file = open("cyb-alignment.fasta", "r")
out_file = open("cyb.phy", "w")
alignment = AlignIO.parse(in_file, "fasta")
AlignIO.write(alignment, out_file, "phylip-relaxed")
in_file.close()
out_file.close()
# Приминение скриптаpython fasta2phy.py
В итоге был получен файл cyb.phy с выравниваниями в нужном формате.
Построение дерева с помощью модели p-distance
Для начала было построено дерево алгоритмом fastme с расчётом расстояний через модель p-distance (число ошибок делить на длину):
fastme -i cyb.phy -o cyb_p_dist.tree -pp-distance
Было получено дерево (рис. 1)
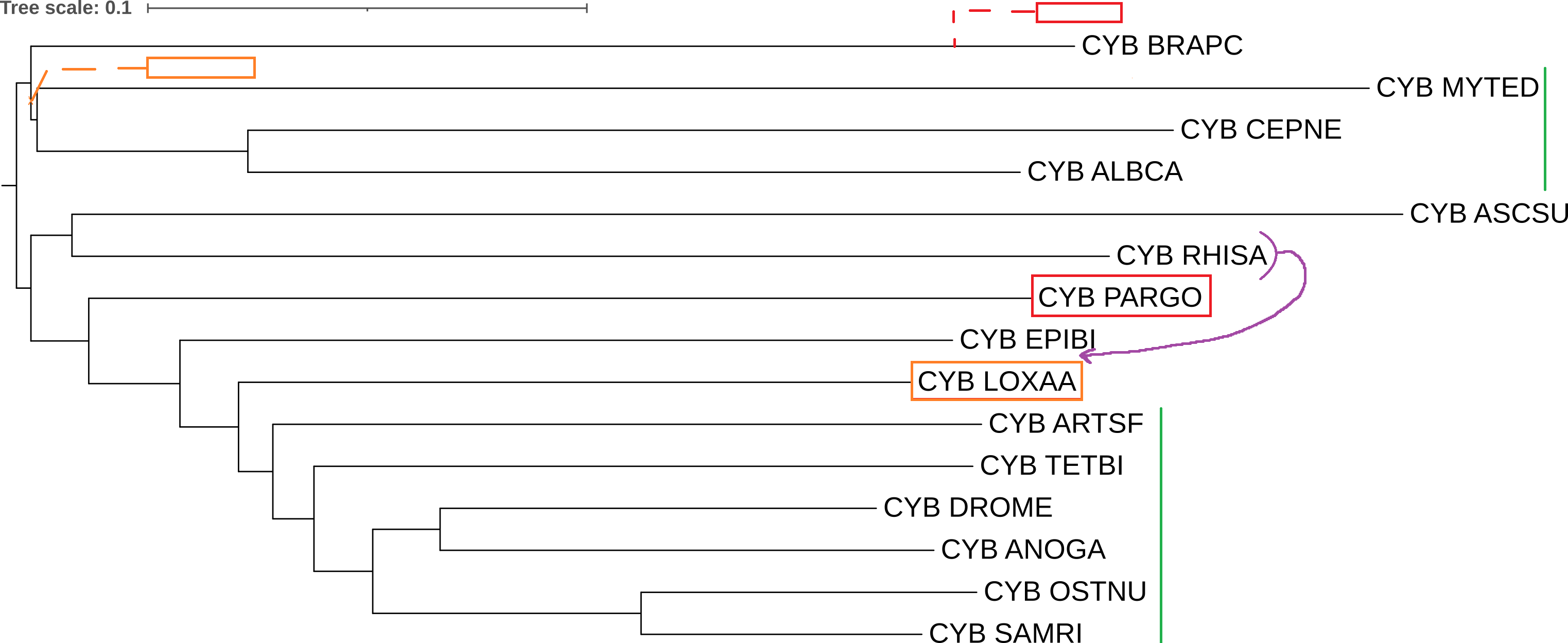
Это дерево похоже на таксономическое, однако есть ряд различий:
- Отсутсвует калада (BRAPC,PARGO). Красным прямоугольником показан организм (PARGO), который оказался не там. Красным пунктиром, заканчивающимся прямоугольником, показано место, где должен располагаться этот организм;
- Организм LOXAA (помечен оранжевым прямоугольником) находится в другой базальной кладе кладе. Оранжевым пунктиром, заканчивающимся прямоугольником, показано место, где должен располагаться этот организм;
- Появилась лишняя клада (ASCSU,RHISA), из-за неверного расположения органима RHISA. Фиолетовым показано, где он должен располагаться;
- Вертикальными зелёными линиями отмечены клады, которые были реконструированы правильно.
Построение дерева с помощью модели MtREV
Модель MtREV (Mitochondrial Reverse) используется для расчёта эволюционных расстояний и построения филогенетических деревьев митохондриальных белков. Она учитывает специфические частоты аминокислотных замен, характерные для митохондрий. Алгоритм построения - fastme.
fastme -i cyb.phy -o cyb_p_dist.tree -pMtREV
Было получено дерево (рис. 2)
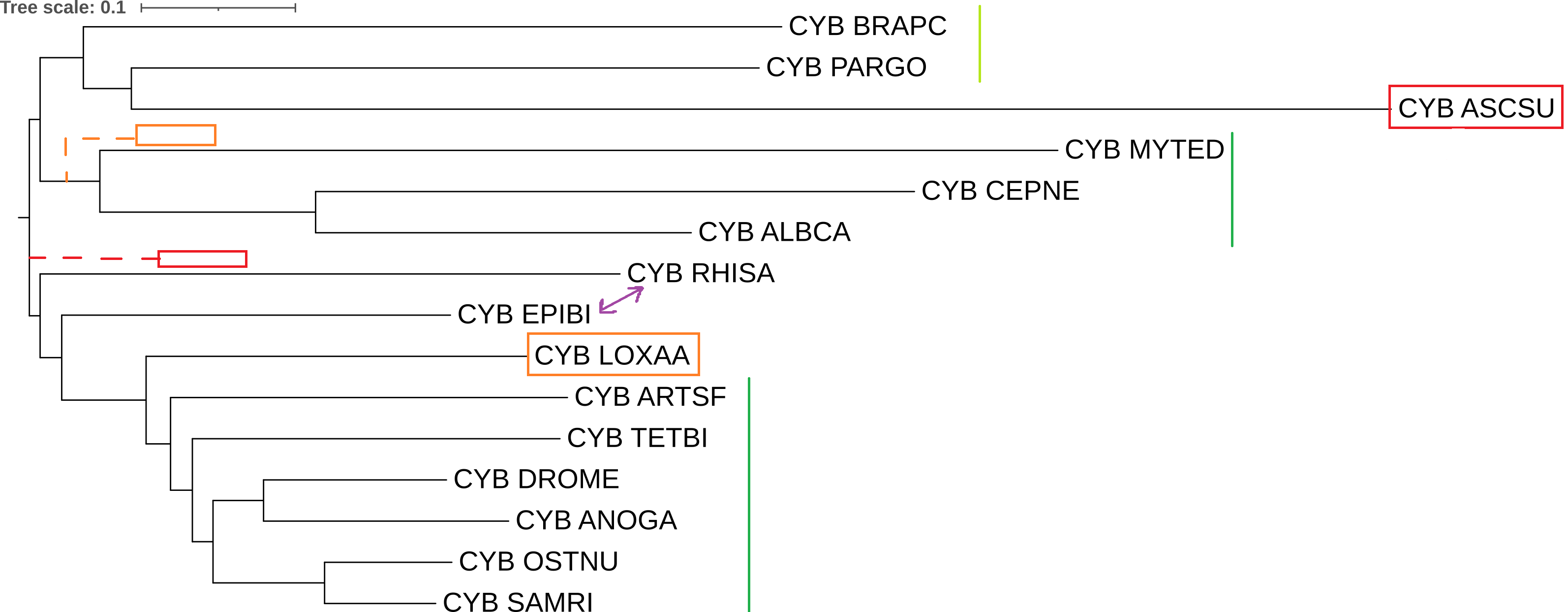
Дерево полученное этим способом кажется более правильным, он также есть неточности:
- Салатовым отмечена "условно" правильная калада (BRAPC,PARGO). "Условно" правильной её было решено сделать в связи с наличием там организма ASCSU. Если бы его не было, токлада была бы верной;
- Организм LOXAA (помечен оранжевым прямоугольником) находится в другой базальной кладе кладе. Оранжевым пунктиром, заканчивающимся прямоугольником, показано место, где должен располагаться этот организм;
- Красным прямоугольником показан организм ASCAU, который оказался не в той базальной кладе и образовал лишнюю кладу (ASCSU,PARGO). Красным пунктиром, заканчивающимся прямоугольником, показано место, где должен располагаться этот организм;
- Фиолетовым показаны организмы, у которых был перепутан порядок (менее базальный стал более базальным);
- Вертикальными зелёными линиями отмечены клады, которые были реконструированы правильно.
Построение дерева с помощью программы IQ-Tree
По умолчанию используется информационный критерий Байеса. Сложняа программа, которая считает расстояния и исходя из некоторых алгоритмов выбтирает наиболее оптимальное.
iqtree -s cyb.phy
Было получено дерево (рис. 3)

Это дерево, по моему мнению, самое неудачное:
- Голубым прямоугольником помечен организм TETBI, который оказался базальнее, чем должен быть. Голубым пунктиром, заканчивающимся прямоугольником, показано место, где должен располагаться этот организм;
- Организм LOXAA (помечен оранжевым прямоугольником) находится в другой базальной кладе вместе с организмом MYTED (синий прямоугольник). Оранжевым и синим пунктирами, заканчивающимися прямоугольниками, показаны места, где должен располагаться эти организмы;
- Красным прямоугольником показан организм PARGO, который оказался не в той базальной кладе и образовал лишнюю кладу (ARTSF,PARGO). Красным пунктиром, заканчивающимся прямоугольником, показано место, где должен располагаться этот организм;
- Фиолетовым показаны организм RHISA, который распалагается слишком базально. Фиолетовым пунктиром, заканчивающимся прямоугольником, показано место, где должен располагаться этот организм;
- Вертикальными зелёными линиями отмечены клады, которые были реконструированы правильно.
Во всех случаях наблюдается проблема с LOXAA и RHISA, а также часто проблемы с PARGO. Интересно изучить, почему это так.