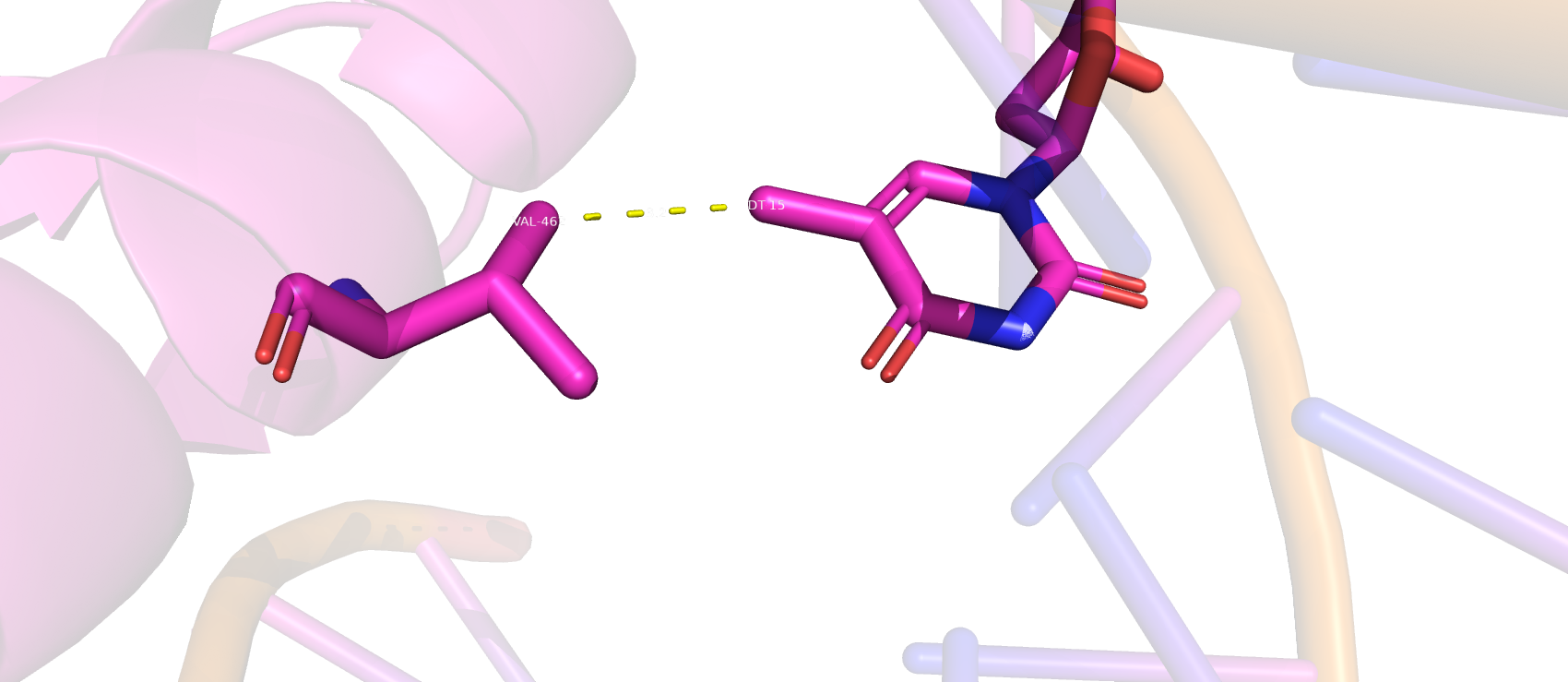По полученной схеме были выбраны Lys461(A) (рис. 1), так как у него самое большое количество связей (3) среди остальных А.О., там более lys461(A) связывается непосредственно с гуанином, что должно увеличивать специфичность распознавания. Также, непосредсвенно с азотистым основанием связывается Val462(A) (рис. 2) одной связью.
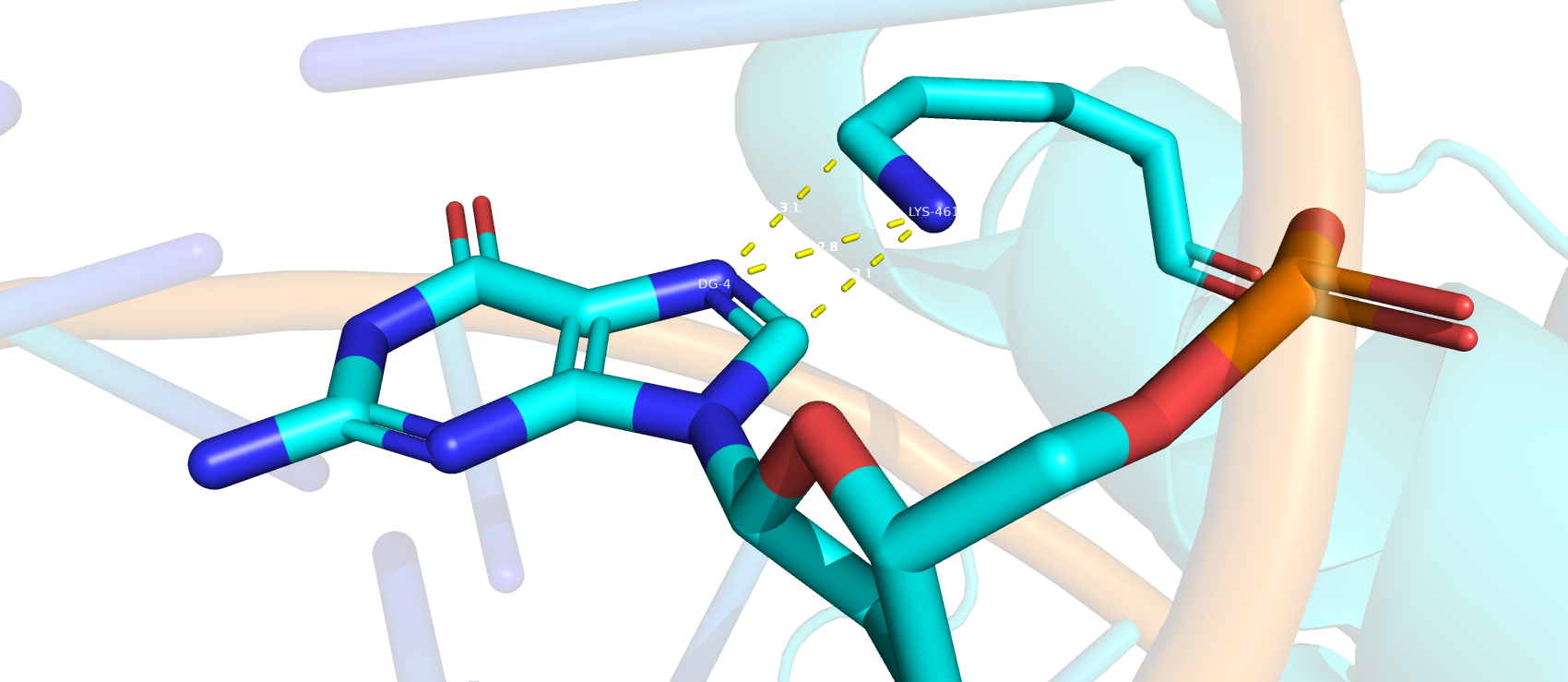
Рисунок 1. Lys461(A) и 3 связи, которые он образует с DG-4¶