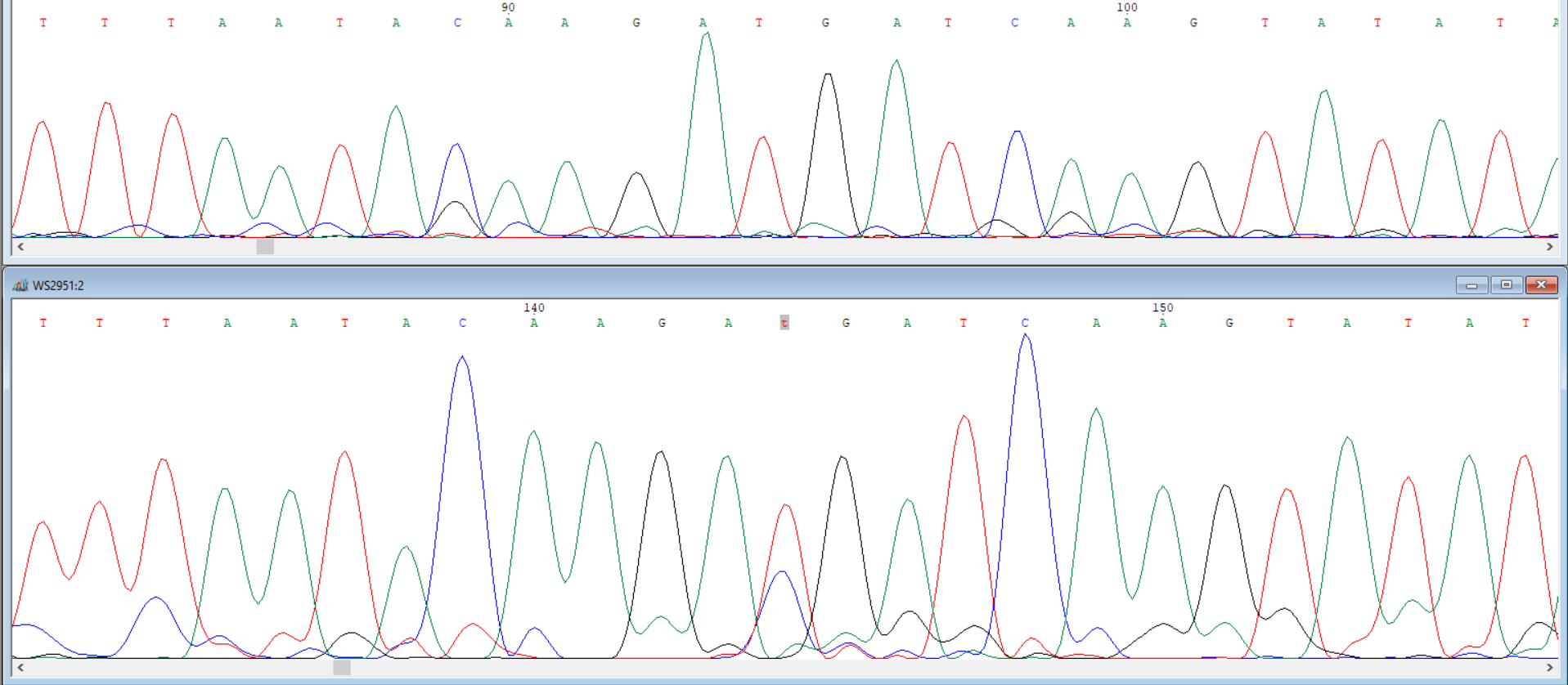
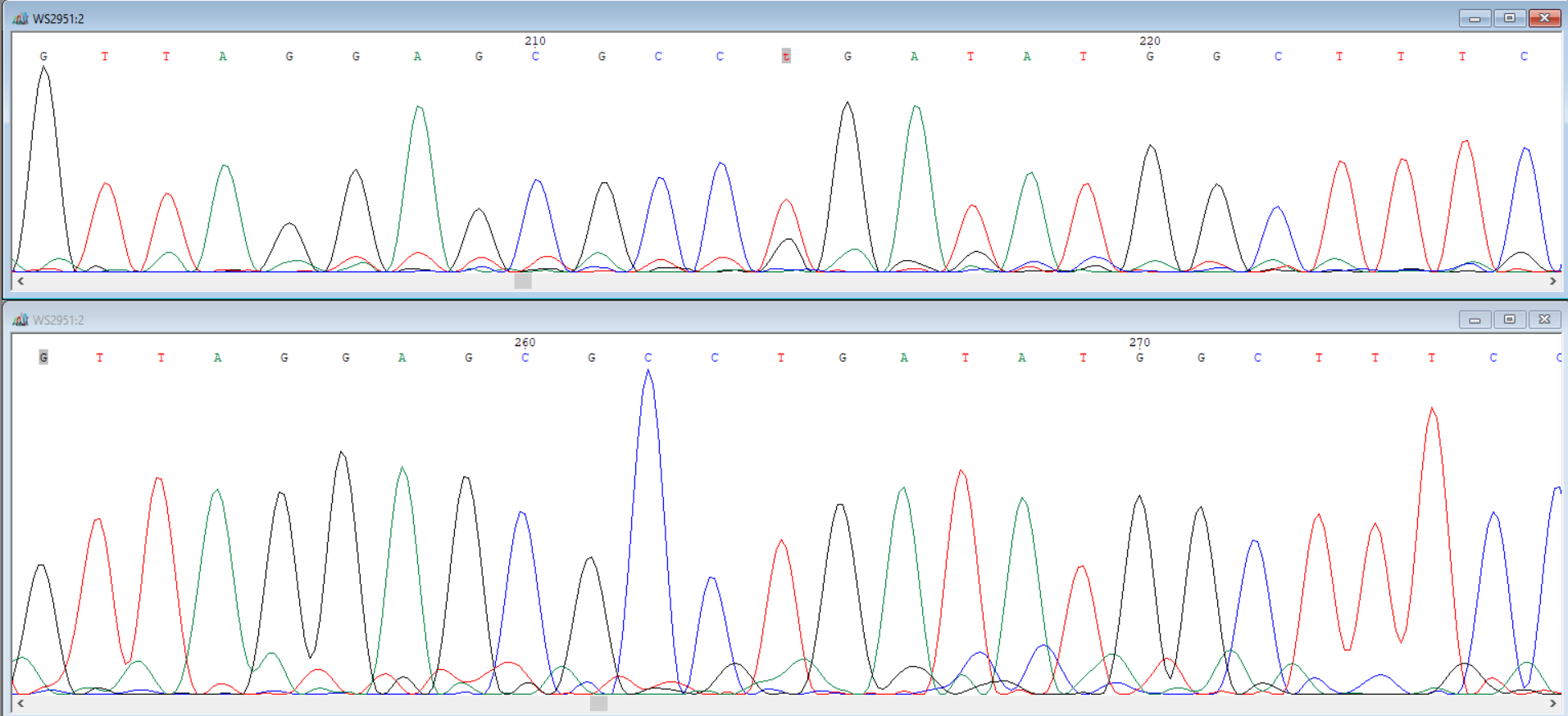
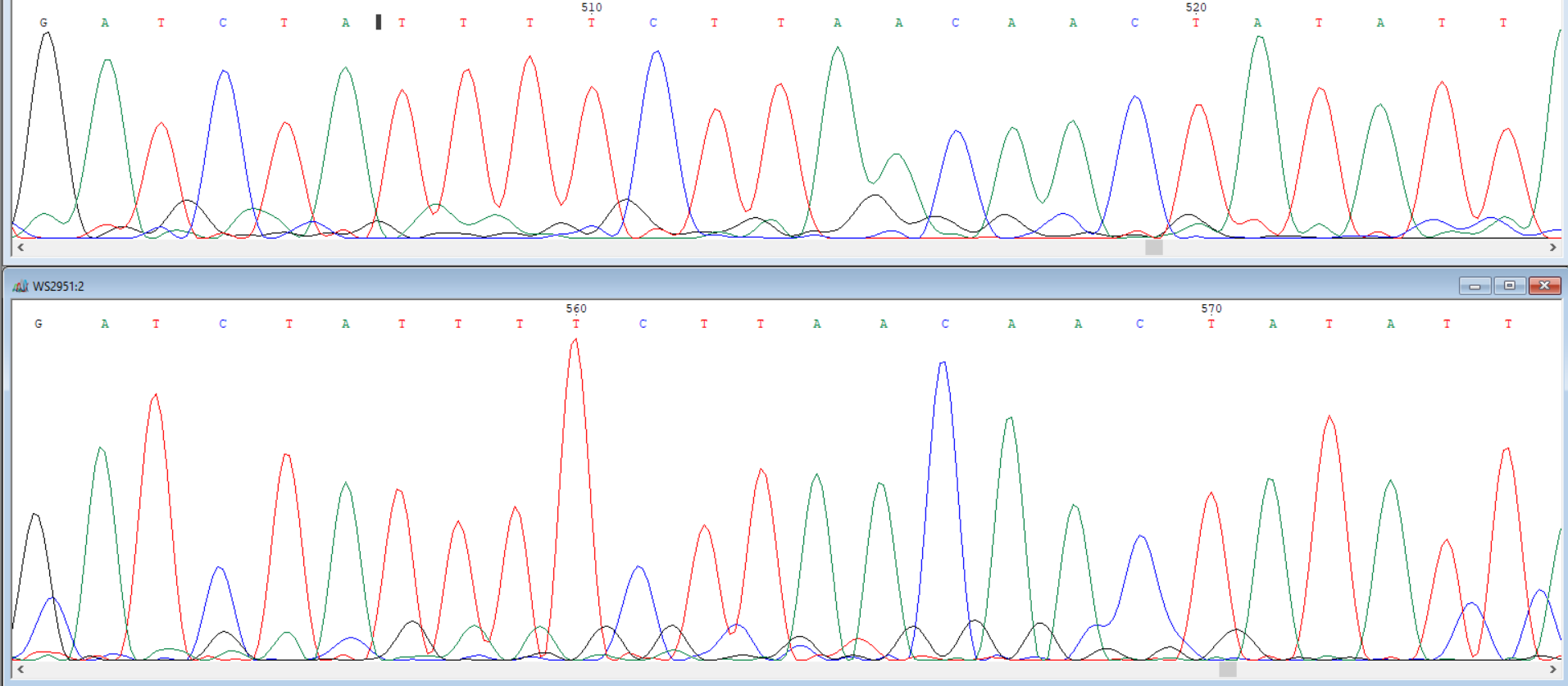
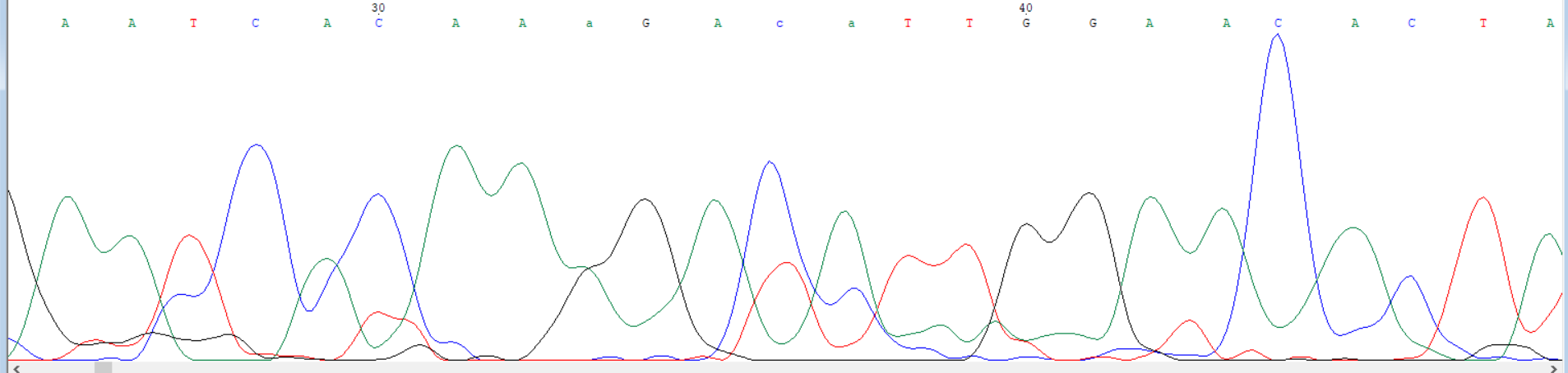
In [3]:
data_quality = {
'name': [],
'position': [],
'quality': []
}
for k in range(len(records)):
for i, n in enumerate(records[k].letter_annotations['phred_quality'], start = 1):
data_quality['position'].append(i)
data_quality['quality'].append(n)
data_quality['name'].append(records[k].name)
data_quality = pd.DataFrame(data_quality)
f, ax = plt.subplots(2, 1, figsize=(14, 8), dpi = 150)
sns.set_context('paper')
sns.lineplot(ax = ax[0], data = data_quality, x = 'position', y = 'quality', linewidth = .7, hue = 'name', ci = 'sd', palette = 'BuPu')
ax[0].set_xlabel('')
sns.lineplot(ax = ax[1], data = data_quality, x = 'position', y = 'quality', linewidth = .7, ci = 'sd', color = '#815DA6')
plt.show()