Evolutional Domains. Profiles
Да простит меня проверяющий: все четыре домашние работы на одной странице (но я сделала оглавление ^_^) :
- Эволюционные домены (часть 1)
- Эволюционные домены (часть 2)
- Построение профиля (pftools)
- Анализ результатов поиска по профилю
Эволюционные домены (часть 1)
Описание баррель-домена пируваткиназы
| AC | PF00224 |
| ID | PK |
| Функция | Баррель-домен пируваткиназы. Этот домен является небольшим доменом типа бета-баррель, расположенным внутри большого TIM-барреля.
Активный центр располагается в щели между двумя доменами. Пируваткиназа (ПК) катализирует последний этап гликолиза (превращение фосфоенолпирувата в пируват, сопровождяющееся фосфорилированием АДФ до АТФ). |
| Число разных доменных архитектур с этим доменом | 22 |
| Число последовательностей | 7047 |
| Число видов | 4703 |
Скачаем выравнивание из Pfam (full) с помощью JalView (File => Fetch). Раскрасим по консервативности (ClustalX => By conservation 10%). Добавим 3D структуру домена к последовательности D8EJP6_ECOLI из PDB ID 1PKY (правая кнопка => Structure => Associate structure with sequence).
Выравнивание в *.fa.
Выравнивание JalView.
Выбор доменных архитектур
С использованием скрипта swisspfam_to_xls.py и файла, содержащего информацию об архитектуре всех последовательностей UniProt (/srv/databases/pfam/swisspfam-2011.gz), была получена таблица с информацией об архитектуре последовательностей, содержащих домен PK, на основе которой в Excel была составлена сводная таблица (лист PivotTable).
python swisspfam_to_xls.py -p PF00224 -i /srv/databases/pfam/swisspfam-2011.gz -z -o swisspfam_to_xls_out.txt
Затем в таблицу были добавлены колонки с информацией о таксономической принадлежности (лист taxonomy). Для этого по идентификаторам отобранных последовательностей в UniProt были получены AC (ID Mapping), по которым были получены файлы в формате UniProt (Retrieve), которые были использованы для работы скрипта uniprot_to_taxonomy.py. Также был добавлен столбец, содержащий информацию о длине доменов PK и PK_C (лист length).
python uniprot_to_taxonomy.py -i retrieved_seq_ids.txt -o uniprot_to_taxonomy_out.txt
Для дальнейшего изучения эволюции доменных архитектур, включающих домен PK, были выбраны архитектуры:
| Архитектура | Число представителей | |
| 1: PK |  |
401 последовательность |
| 2: PK + PK_C* |  |
3451 последовательность |
Описание домена PK_C
| AC | PF02887 |
| ID | PK_C |
| Функция | Альфа/бета-домен пируваткиназы. Наряду с тем, что является компонентом пируваткиназ, обнаружен также как самостоятельный домен в некоторых белках бактерий. |
| Число разных доменных архитектур с этим доменом | 21 |
| Число последовательностей | 6414 |
| Число видов | 4576 |
Выбор таксона и представителей архитектур
Были выбраны два больших таксона Bacteria и Eukaryota.
Представители были выбраны при работе с таблицей (лист PivotTable, а уже выбранные последовательности лист SelectedSeqs).
Для каждой архитектуры было отобрано по 24 последовательности (по 12 из каждого таксона). Чтобы оставить в выравнивании нужные последовательности из двух групп (был использован скрипт filter_alignment.py).
python filter_alignment.py -i pk.fa -m selected_ids.txt -o filter_alignment_out.fa
Полученное выравнивание было загружено в JalView и отредактировано (удалены пустые колонки). Затем в нём были выделены группы согласно архитектуре, в каждой из них была выполнена раскраска последовательностей ClustalX, Conservation (порог на консервативность 10%). После повторного редактирования выравнивания (удалены несодержательные N- и C-концевые участки, удалены некоторые последовательности, снова удалены пустые колонки) в файле выравнивания JalView содержится 38 последовательностей.

Таблица Excel (листы swisspfam_to_xls.out --- SelectedSeqs).
Эволюционные домены (часть 2)
Построение филогенетического дерева выборки последовательностей домена PK
Зашифровка доменных архитектур и сравниваемых таксонов в названиях последовательностей:
| Архитектура | 1 | Архитектура №1 (PK) |
| 2 | Архитектура №2 (PK + PK_C) | |
| Таксон | B | Bacteria |
| E | Eukaryota |
Выбранные последовательности (выравнивание selected.fa). Все деревья были графически построены в ITOL.
Построим филогенетическое дерево выбранных последовательностей, используя известные программы. В деревьях, построенных с помощью программ fprotpars (метод maximal parsimony, img) и Average Distance (метод UPGMA из JalView, img), установить какие-либо закономерности эволюции домена не удается.
В дереве, сконструированном программой Neighbor Joining (метод NJ из JalView, img), более-менее прослеживается ветвь (синий), содержащая эукариотические последовательности (красные листья):
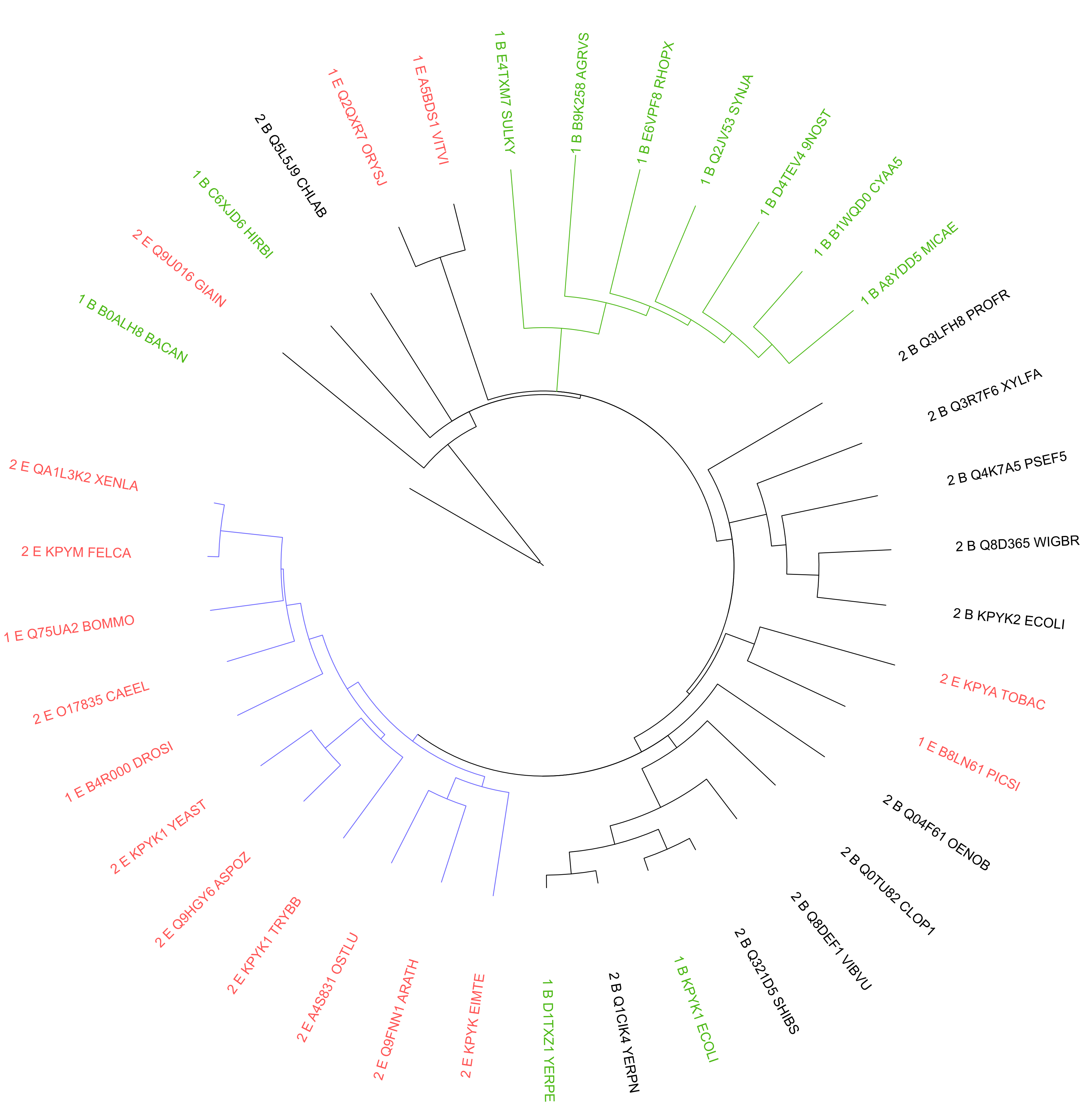
Укоренение дерева, посроенного методом NJ, в среднюю точку с помощью программы retree результатов, которые можно пообсуждать.
Кажется вероятной независимая эволюция доменных архитектур у представителей бактерий и эукариот.
Бактериальные последовательности из однодоменной архитектуры выделяются в обособленную ветвь на дереве, построенном методом NJ (зеленый). Соответственно можно представить, что из ранее существующей у бактерий двухдоменной архитектуры каким-то образом произошло обособление этого домена. Эукариотические последовательности не образуют клад, коррелирующих с доменной архитектурой. Также ничего нельзя сказать о том, какая из архитектур была представлена у общего предка эукариот.
Построение профиля (pftools)
Создание профиля
Был использован файл selected.msf, сохраненный из JalView. Затем он был отредактирован с помощью следующих команд (добавлены в выравнивание веса последовательностей (программа pfw) и создан профиль (команда pfmake строит профиль PROSITE на основании множественного выравнивания)):
#приведение к правильному формату файла seqret selected.msf msf::selected1.msf noreturn -infile selected1.msf -outfile selected2.msf #добавление весов в выравнивание pfw selected2.msf > selected_weighted.msf #создание профиля pfmake selected_weighted.msf /usr/share/pftools23/blosum62.cmp > selected.prf
Нормализация профиля
Нормализацию профиля selected.prf можно выполнить с использованием банка случайных последовательностей (/srv/databases/uniprot/sprot_shuffled.fasta, полученный перемешиванием каждой из последовательностей swissprot).
#команда поиска по профилю pfsearch -C 40 -f selected.prf /srv/databases/uniprot/sprot_shuffled.fasta | sort -nr > selected_scores.txt
Замечание 1. Опция -C с целым значением задает порог на обычный вес. Опция -C с десятичным значением - с точкой - задает порог на нормализованный вес.
Замечание 2. Опция -C 10 отрезает случайные находки с весом меньше 10, уменьшая размер выдачи. Если находок остается достаточное число
- несколько тысяч, то их достаточно для нормализации.
#команда нормализации профиля pfscale selected_scores.txt selected.prf > selected_scaled.prf
Поиск по профилю в банке SwissProt
Для поиска по банку SwissProt был использован нормализованный профиль selected_scaled.prf, при этом порог нормализованного веса был установлен равным 500.0 (default = 8.5). Ожидается, что находки с нормализованным весом более, чем мы устанавливаем - правильные
#команда поиска по профилю в SW pfsearch -C 5.5 -f selected_scaled.prf /srv/databases/uniprot/sprot.fasta | sort -nr > selected_result.xlsТак как с порогом 5.5 находок очень много (552983), то я решила подобрать порог нормализованного веса для поиска по банку SwissProt - 150:
#команда поиска по профилю в SW pfsearch -C 150.0 -f selected_scaled.prf /srv/databases/uniprot/sprot.fasta | sort -nr > selected_result_150.xls
В полученном файле selected_result_150.xls содержится более 2652 последовательности, что достаточно для дальнейшего построения графика весов находок и ROC-кривой таблица Excel (листы selected_result_150, selected_roc.
Анализ результатов поиска
таблица Excel лист selected_result_150.
Отметим в талице последовательности, в которых имеется домен Pfam. Список последовательностей, содержащих данный домен, был получен при выполнении предыдущих заданий.(см. Эволюционные домены (часть 1) => Выбор доменных архитектур, таблица Exel лист swisspfam_to_xls_out).
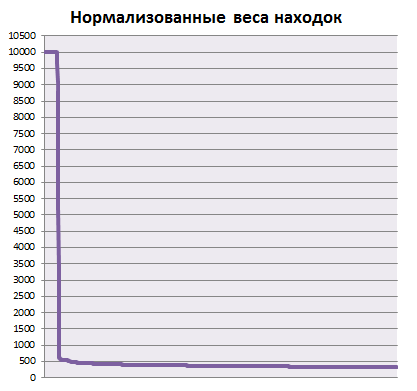
Построим график весов находок pfsearch, отсортированных по убыванию. Ступенька на этом графике можно интерпретировать как порог нормализованного веса для находок из семейства. Средствами Excel для полученного списка находок был построен график нормализованного веса находок, «ступеньку» на котором можно интерпретировать как порог нормализованного веса для находок из семейства (в данном случае равен 150):
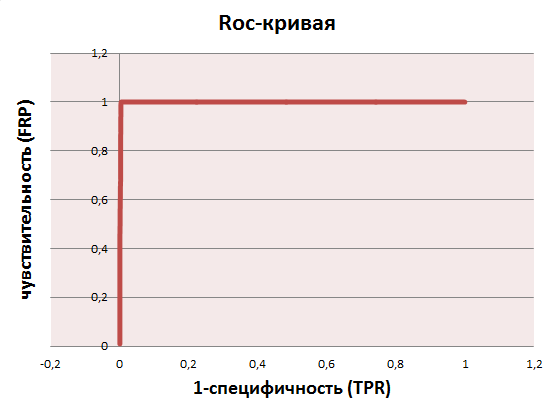
ROC-кривая представляет собой зависимость чувствительности алгоритма классификации (true positive rate, TPR) от величины FPR (false positive rate), которую можно обозначить как 1 – специфичность. Построенная в Excel ROC-кривая по полученному списку находок (таблица Excel (лист selected_roc):
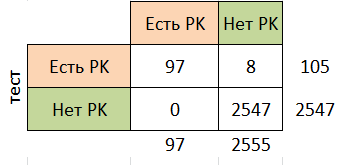
На основе данных, приведённых на листе selected_result_150, можно установить порог нормализованного веса, равный 2053, который даёт 0 ошибок I рода (0%; это число соответствует вероятности не определить последовательность, содержащую домен PK) и 8 ошибок II рода (7%; это значение соответствует вероятности определить последовательность, не имеющую домена PK, как принадлежащую семейству).
Анализ результатов поиска по профилю
Разделение выравнивания на две группы
Разделим выравнивание на две группы по таксономии организмов, поскольку на дереве, построенном методом NJ, можно проследить некоторое соответствие таксономическим группам (см. картинку ранее).
Построение профиля, отличающего бактериальные последовательности от эукариотических
Выравнивания, используемые далее для создания профиля: b.msf (содержит все бактериальные последовательности из выравнивания) и e.msf (все эукариотические последовательности минус те, о которых я скажу ниже).
seqret e.msf msf::e1.msf noreturn -infile e1.msf -outfile e2.msf pfw e2.msf > e_weighted.msf pfmake e_weighted.msf /usr/share/pftools23/blosum62.cmp > e.prf
Далее был создан fasta-файл со всеми 38 последовательностями, пережившими редактирование выравнивания JalView. Но было обнаружено,что последовательности >2_E_QA1L3K2_XENLA каким-то чудесным образом вообще нет в Uniprot, а последовтельность >1_E_B4R000_DROSI представления своим фрагментом. Поэтому было принято решение о том, чтобы исключить их из файлов для построения профилей. Обе последовательности эукариотические, поэтому был переделан профиль e.prf, а профиль b.prf оставлен неизменным. fasta-файла со всеми 36 последовательностями, пережившими все эти редактирования. Последовательности все еще зашифрованы (см. расшифровку выше).
Поиск по профилю в исходных последовательностях
Далее, проведем cам поиск по профилю в исходных последовательностях. Порог веса поставим маленьким (-C 0.0), чтобы все последовательности оказались в выдаче.
#команды поиска по b и e профилям в исходных последовательностях pfsearch –C 0.0 –f b/b.prf selected_seqs.fa | sort -nr > b.xls pfsearch –C 0.0 –f e/e.prf selected_seqs.fa | sort -nr > e.xls
Результаты поиска при помощи полученных профилей по исходным последовательностям - Таблица Excel (листы b, e).
Построение ROC-кривой по полученным данным и анализ результатов поиска
таблица Excel листы b, roc_b, e, roc_e.
Отметим в талицах последовательности, которые действительно принадлежат профилям. Построим график весов находок pfsearch, отсортированных по убыванию.
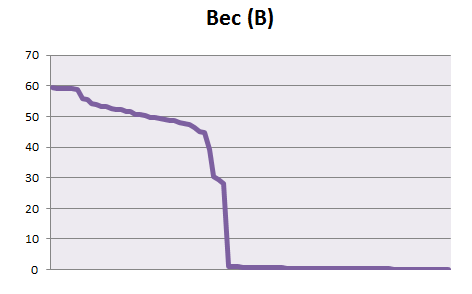
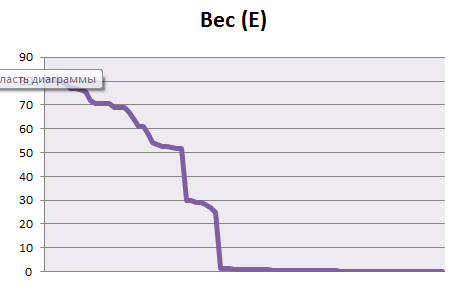
ROC-кривые:

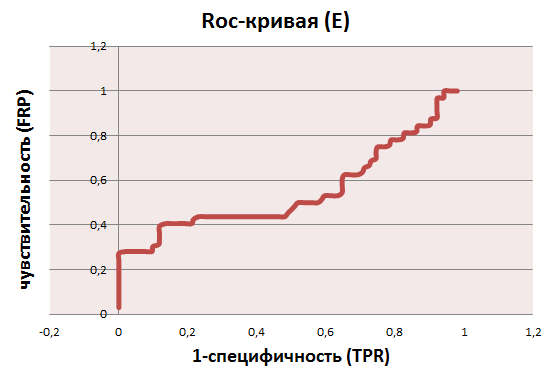
На основе данных, приведённых на листах b и е, можно установить порог нормализованного веса, равный 44 для бактериального профиля и 24 для эукариотического. Пороги установлены по "скачкам" весов и по столбцу принадлежности, однако, при таких порогах мы все равно имеем большие проценты ошибок I и II рода:
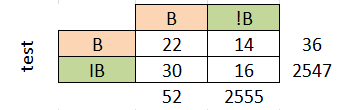
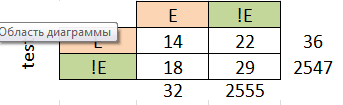
Это говорит о том, что по таким построенным профилям различать эукариотические и бактериальные последовательности лучше не стоит, т.к. маленькая достоверность.
Наверх