Phylogenetic Trees Reconstruction
Задание 1.
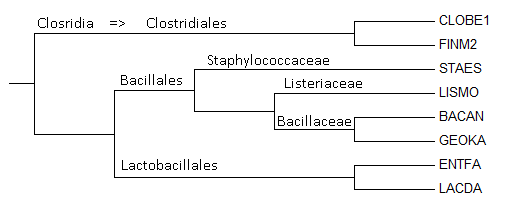
Пользуясь таксономическим сервисом NCB, определить, к каким таксонам относятся отобранные в предыдущем задании бактерии. Определить, есть ли на дереве отобранных бактерий ветви, выделяющие какие-нибудь из таксонов.Ясно, что все выбранные бактерии относятся к Cellular organisms (клеточные организмы) и Bacteria (Бактерии). Кроме того, они все Firmicutes (грамположительные).
| Таксоны | Название | Мнемоника |
| Bacilli; Bacillales; Bacillaceae; Bacillus; Bacillus cereus group | Bacillus anthracis | BACAN |
| Clostridia; Clostridiales; Clostridiaceae; Clostridium | Clostridium botulinum | CLOB1 |
| Clostridia; Clostridiales; Clostridiales incertae sedis; Clostridiales Family XI. Incertae Sedis; Finegoldia | Finegoldia magna | FINM2 |
| Bacilli; Lactobacillales; Enterococcaceae; Enterococcus | Enterococcus faecalis | ENTFA |
| Bacilli; Bacillales; Bacillaceae; Geobacillus | Geobacillus kaustophilus | GEOKA |
| Bacilli; Lactobacillales; Lactobacillaceae; Lactobacillus | Lactobacillus delbrueckii | LACDA |
| Bacilli; Bacillales; Listeriaceae; Listeria | Listeria monocytogenes | LISMO |
| Bacilli; Bacillales; Staphylococcaceae; Staphylococcus | Staphylococcus epidermidis | STAES |
Задание 2.
Получим из Swiss-Prot последовательности белков с функцией шаперонина (HSLO) из отобранных бактерий:seqret sw:hslo_bacanПоместим последовательности в один fasta-файл all.fasta (раз уж нас начали учить python'у, то с помощью скрипта) и отредактируем названия последовательностей, оставив только мнемонику видов.
Создадим выравнивание отобранных белков. Для этого на kodomo запустим программу muscle с параметрами по умолчанию (т.к. нам не сказано, кк выравнивать) и затем откроем это выравнивание в JalView.
muscle -in all.fasta -out align.fastaПолучается такая картинка выравнивания (блочная форма (wrap), раскраска по проценту идентичности (percentage identity)):

Проект JalView выравнивания и выравнивание в fasta-формате.
Задание 3.
Сложно выделить какие-нибудь диагностические позиции выравнивания (уж очень разноцветное выравнивание), однако все-таки встречаются столбцы, выделающие- Clostridia от остальных: 30-31, 33, 37, 61, 75, 114, 153, 169, 176.
- Lactobacillales (LACDA, ENTFA): 21, 39, 49, 109.
Задание 4.
Реконструируем филогенетическое дерево четырьмя методами, доступными из JalView (меню Calculate → Calculate tree). Каждое дерево сохраним в Newick-формате в файл с соответствующим названием, а в отчёте - изображения деревьев, сделанные программой Mega.Сравнение топологии полученных деревьев с топологией правильного дерева
Для каждого из четырёх вариантов укажите, какие ветви есть в дереве, построенном JalView, но отсутствуют в правильном, и какие — наоборот.
Во всех деревьях (BACAN,GEOKA) вынесены в одну ветвь, что говорит о близком сходстве их шаперонина HSLO.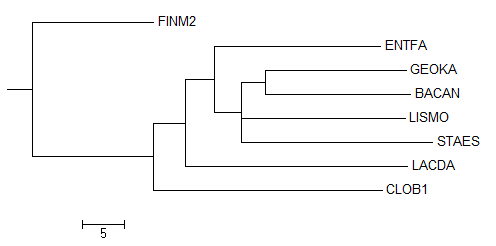
1) Neigbour joining tree using PID
Кажется, что дерево не выделяет Clostridia (CLOB1 AND FINM2) в отдельную ветвь, но на самом деле этот метод не укореняет дерево нигде. Поэтому ясно, что укоренить можно и в правильном месте вот так:((FINM2,CLOB1),(...)).
Ветвь Bacillales (STAES+LISMO+BACAN+GEOKA) отделена верно, но положение STAES не соответсвует правильному:(STAES,LISMO,(BACAN,GEOKA)), [верно - (STAES,(LISMO,(BACAN,GEOKA)))].
ENTFA не выделена в отдельную ветвь с LACDA: (LACDA,(ENTFA,(...))) [верно - ((LACDA,ENTFA),(...))]
Верные разбиения (нетривиальные ветви):- {BACAN,GEOKA} vs {CLOT1,FINM2,STAES,LISMO,LACDA,ENTFA};
- {CLOT1,FINM2} vs {BACAN,GEOKA,STAES,LISMO,LACDA,ENTFA};
- {BACAN,GEOKA,STAES,LISMO} vs {CLOT1,FINM2,LACDA,ENTFA}.
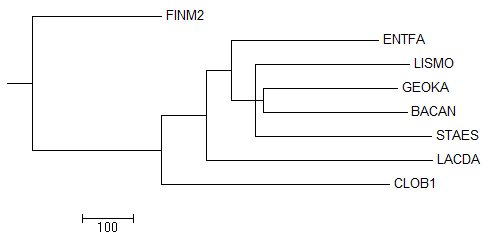
2) Neigbour joining tree using BLOSUM62
Повторяет дерево 1), за исключением топологии ветви Bacillales (STAES,(LISMO,(BACAN,GEOKA))), но все равно похоже.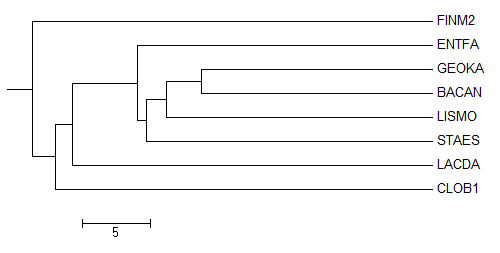
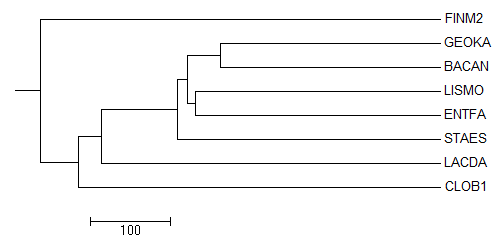
3) Average distance tree using PID
Это дерево уже действительно не выделяет Clostridia (CLOB1 AND FINM2) в отдельную ветвь: (FINM2, (...)), в то время как правильно ((FINM2,CLOB1),(...)).
ENTFA не выделена в отдельную ветвь с LACDA: (LACDA,(ENTFA,(...))) [верно - ((LACDA,ENTFA),(...))]
Верные разбиения (нетривиальные ветви):- {CLOBE1,FINM2} vs {STAES,LISMO,BACAN,GEOKA,ENTFA,LACDA};
- {GEOKA,BACAN} vs {CLOBE1,FINM2,STAES,LISMO,ENTFA,LACDA};
- {GEOKA,BACAN,LISMO} vs {CLOBE1,FINM2,STAES,ENTFA,LACDA};
- {GEOKA,BACAN,LISMO,STAES} vs {CLOBE1,FINM2,ENTFA,LACDA}.
4) Average distance tree using BLOSUM62
Дерево также действительно не выделяет Clostridia (CLOB1 AND FINM2) в отдельную ветвь: (FINM2, (...)), в то время как правильно ((FINM2,CLOB1),(...)).
ENTFA не выделена в отдельную ветвь с LACDA,а вместо этого пристроена в одну ветвь с LISMO в большой ветви Bacillales (LACDA,(STAES,((LISMO,ENTFA),(BACAN,GEOKA)))) [верно - ((LACDA,ENTFA),(STAES,(LISMO,(BACAN,GEOKA))))]
Верные разбиения (нетривиальные ветви):- {CLOBE1,FINM2} vs {STAES,LISMO,BACAN,GEOKA,ENTFA,LACDA};
- {GEOKA,BACAN} vs {CLOBE1,FINM2,STAES,LISMO,ENTFA,LACDA};
Задание 5.
Импортируем выравнивание в программу Mega (при импорте - "Analyze"). Реконструируем дерево методом "Maximum Parsimony" (из Phylogeny). Укореним дерево так, чтобы ветвь Clostridia была противопоставлена всему остальному (Subtree → Root):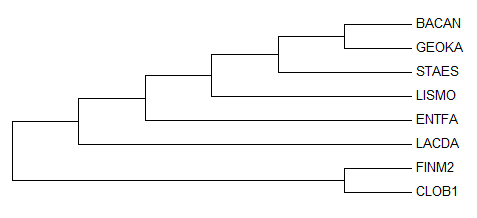
Дерево отличается от правильного тем, что STAES "роднее" (BACAN,GEOKA), чем LISMO, а также тем, что ENTFA не выделена в отдельную ветвь с LACDA: (LACDA,(ENTFA,(...))) [верно - ((LACDA,ENTFA),(...))]
Наверх