d1. Определение вторичной структуры
Для работы была выбрана структура 1BAM - эндонуклеаза рестрикции BamHI. В PDB-файле модели этой структуры уже указана разметка вторичной структуры - 7 альфаспиралей и 7 бета-листов:
HELIX 1 A1 ASP A 10 LEU A 16 1 7
HELIX 2 A2 LYS A 20 CYS A 34 1 15
HELIX 3 A3 VAL A 58 ASP A 70 1 13
HELIX 4 A4 ILE A 117 LYS A 132 1 16
HELIX 5 A5 LYS A 146 ALA A 149 1 4
HELIX 6 A6 PHE A 159 THR A 169 1 SEE REMARK 6. 11
HELIX 7 A7 LYS A 200 GLU A 211 1 12
SHEET 1 SA 6 TYR A 75 ARG A 76 0
SHEET 2 SA 6 VAL A 95 GLU A 101 -1
SHEET 3 SA 6 GLU A 104 PHE A 112 -1
SHEET 4 SA 6 ASP A 137 PRO A 144 1
SHEET 5 SA 6 PHE A 174 ASN A 180 1
SHEET 6 SA 6 GLU A 2 ILE A 8 -1
SHEET 1 SB 2 THR A 46 ASN A 48 0
SHEET 2 SB 2 ALA A 183 ASN A 185 1
REMARK 650 HELIX
REMARK 650 HELIX A6 IS A MIXTURE OF TYPE 1 AND TYPE 5. PRO 164 IN THE
REMARK 650 MIDDLE OF THE HELIX KINKS IT. RESIDUES 159 - 162 FORM AN
REMARK 650 ALPHA HELIX. RESIDUE 163 - 165 AND 166 - 169 FORM A 3/10
REMARK 650 HELIX.
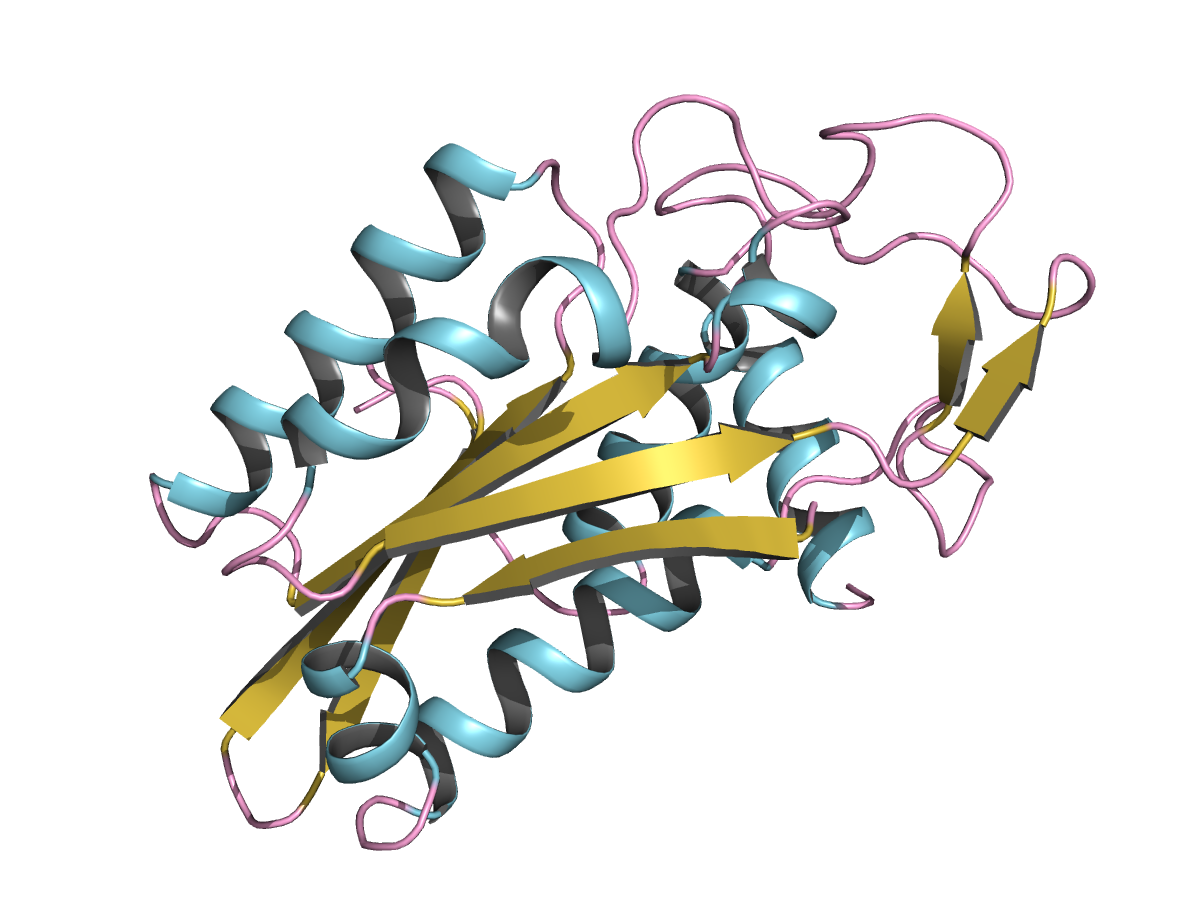 |
| Рис.1. Общий вид структуры 1BAM |
DSSP
DSSP был запущен на kodomo:
mkdssp -i 1BAM.pdb -o 1BAM.dssp
Результат работы алгоритма представлен в файле 1BAM.dssp.
Координаты спиралей и листов определились достаточно точно. Несовпадение с разметкой PBD-файла только в одной спирали и в одном листе, но только на 1 и 2 остатка:
138-144 <= SHEET 4 SA 6 ASP A 137 PRO A 144 1
58-72 <= HELIX 3 A3 VAL A 58 ASP A 70 1
Кроме того, программа правильно определила особенность спирали HELIX: PHE159 - THR169, в том, что со 163 остатка это 3/10 спираль (в PDB-файле эта особенность тоже отмечена и объяснена - PRO164 изгибает спираль). Так же в структуре были найдены искривленные участки (bends) и повороты полипептидной цепи. Еще обнаружились такие изолированные бета-тяжи (В), которые состоят из одиночных остатков, что вообще-то не очень верно.
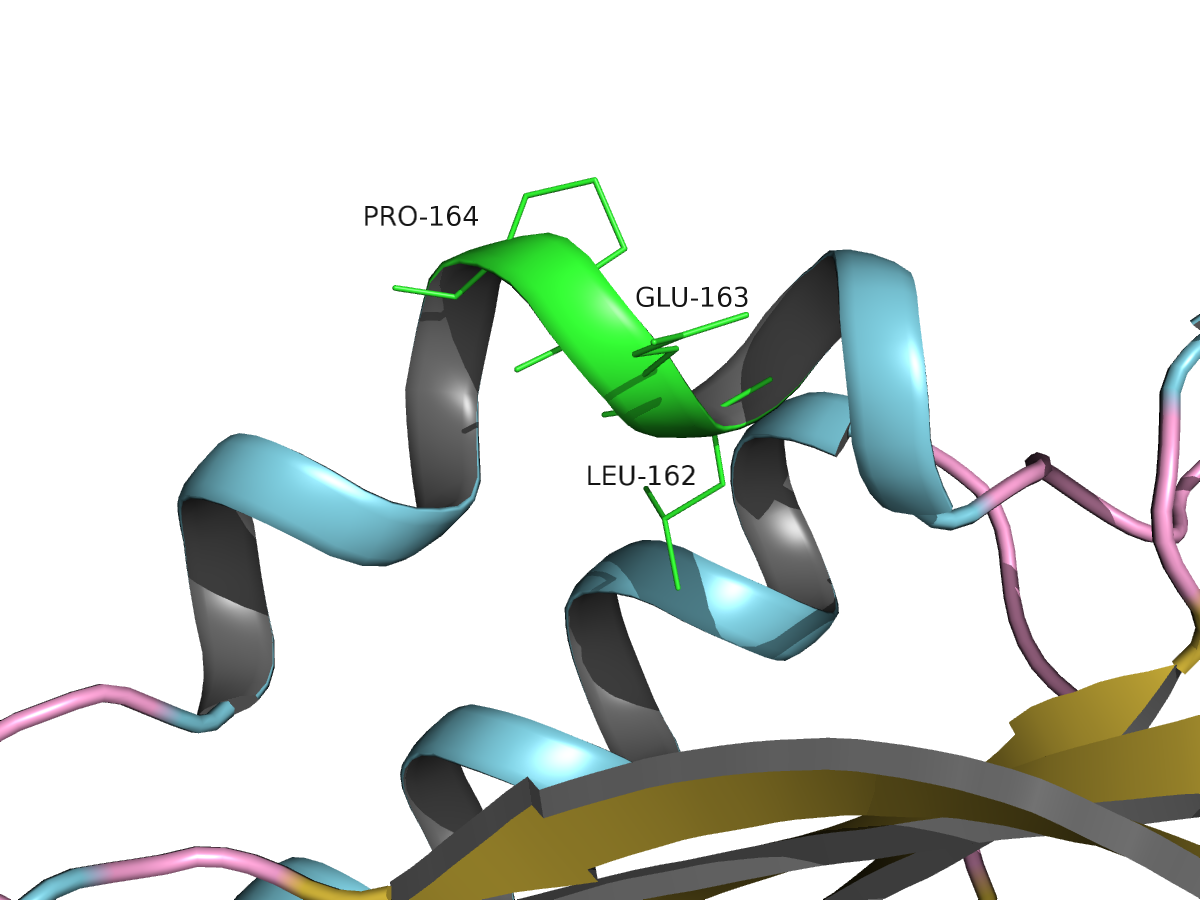 |
| Рис.2. Изгиб спирали под влиянием PRO164 |
SheeP
Структура 1BAM была загружена на веб-сервис для построения карт бета-листов SheeP.
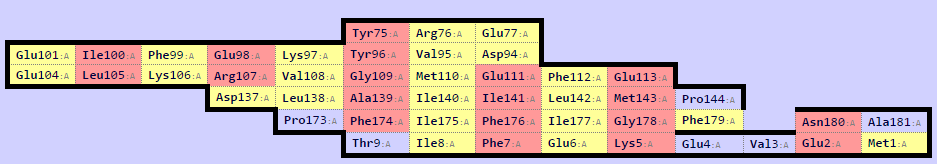 |
| Рис.3. Карта одного из бета-листов структуры (самого большого), полученная при помощь SheeP |
На карте видно, что это один большой бета-лист. На Рис. 4 показано расположение этого бета-листа во всей структуре.
Так как струтура представляет собой сэндвич, сложно сказать, где гидрофильная сторона, а где гидрофобная.
Все остальные бета-тяжи определены более менее точно, кроме того, что представлен на Рис.5 (все определенные SheeP листы - в проволочной модели,
розовые петли по разметке, желтые тяжи по разметке, голубые - спирали).
 |
| Рис.4. Структура 1BAM с показанным самым большим бета-листом цепи (жирные линии). Серым и красным отмечены атомы, торчащие в разные стороны от листа. |
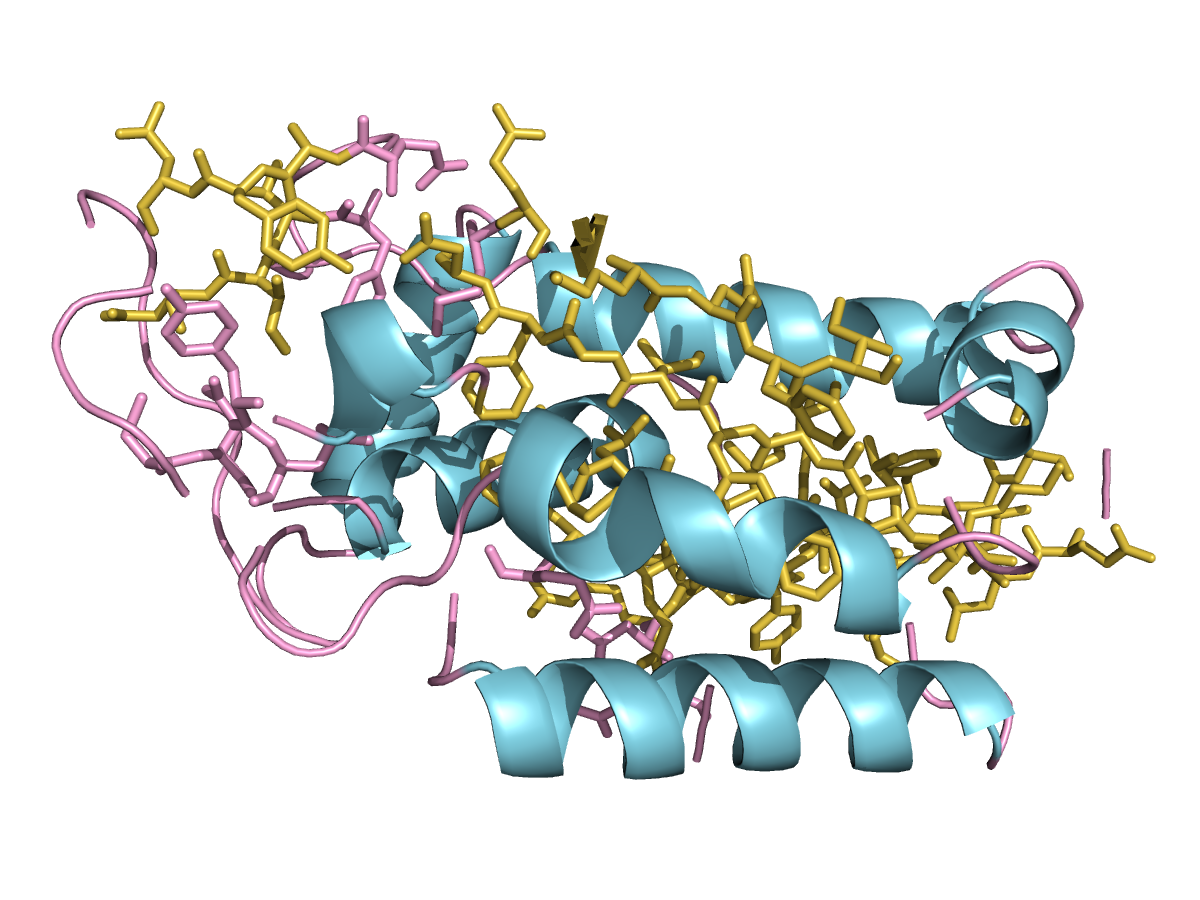 |
| Рис.5. Ошибочное определение бета-листа. Дополнительный бета-лист (розовая проволочная - должна быть петля, а там тяжи). |
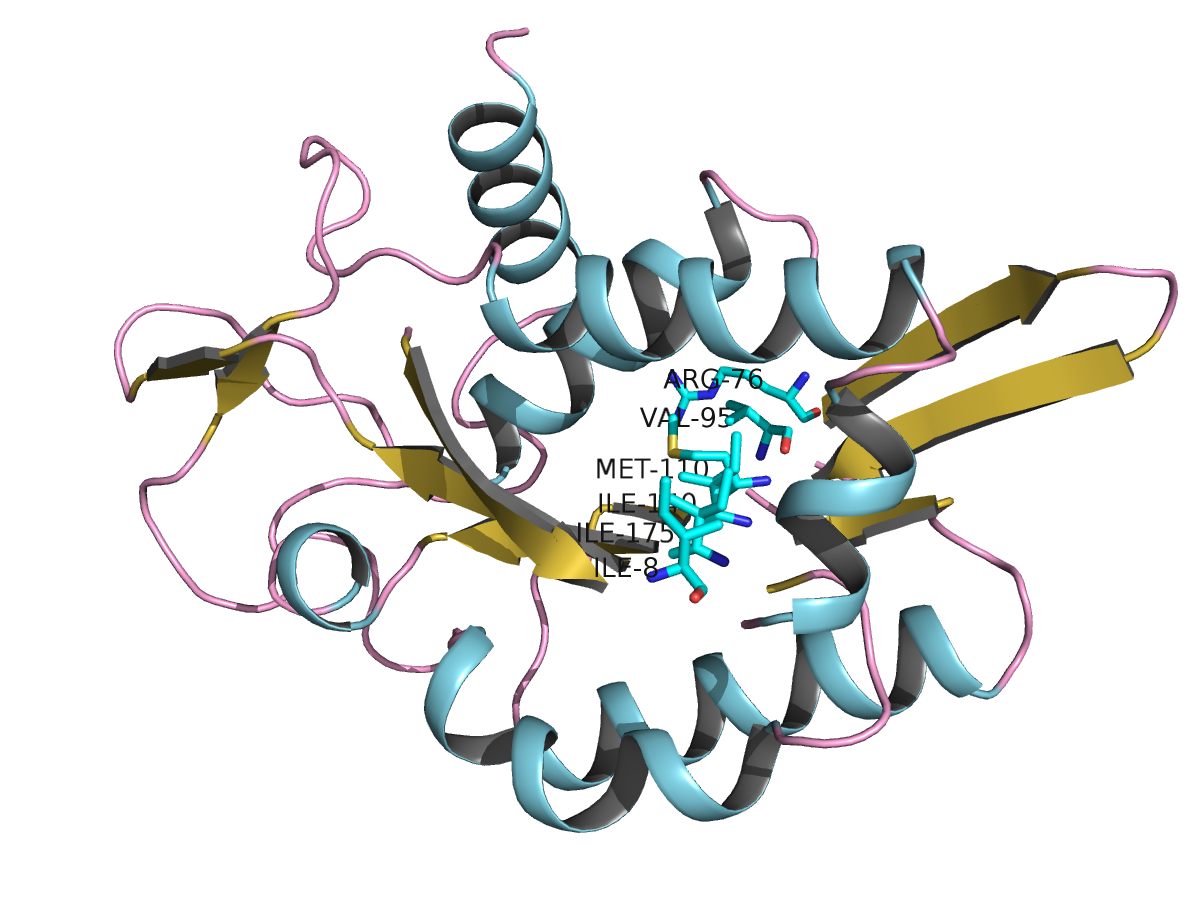 |
| Рис.6. Самый длинный хребет бета-листа: остатки 76,95,110,140,175,8 цепи А предствалены в виде остова, совпадает с разметкой PDB-файла. |
d2. Совмещение структур
Совмещение структуры кроличьей ГАФД с ее структурными гомологами
Cтруктурные гомологи белка были найдены при помощи сервиса PDBeFold => launch => pairwise 3D alignment, query - PDB код, target – whole pdb archive, поиск структурных гомологов для всех цепей, lowest acceptable match по умолчанию 70%.
В таблице с находками было выбрано 6 разных белков с RMSD (оценка сходства) от 0,8 до 2,5 ангстрем и N_align(число сопоставленных C_alpha) от 50% до 90%. Сервис сам показывает картинку совмещения структур(Рис. 7). Можно сказать, что структуры в первом приближении очень похожи.
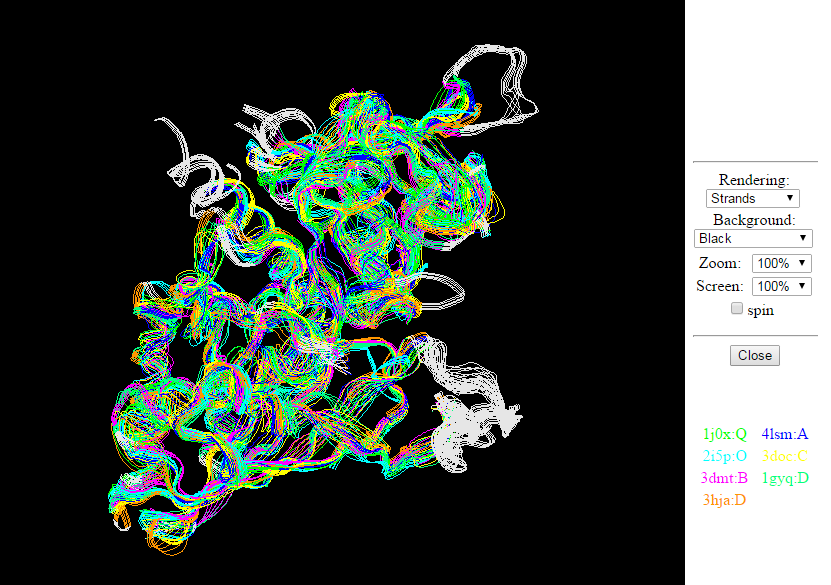 |
| Рис.7. Совмещение структуры 1J0X и ее пяти структурных гомологов. |
Выравнивание последовательностей по совмещению структур; совмещение структур.
Выравнивание последовательностей Tcoffee.
Рассмотрим петлю 3hja:D 102-105. В структурно выравнивании она не совпадает с другими структурами, что видно на картинке (Рис.8). Из выравниваний (Рис.9) видно, что аминокослоты надо 106-109 для последней последовательностью плохо выравниваются с любым участком из куска 102-109, поэтому все равно куда их плохо выровнять. Поэтому сложно сказать, где правильное выравнивание. В принципе, выравнивания очень похожи.
 |
| Рис.8. Совмещение структуры 1J0X и ее пяти структурных гомологов, выпетливание невыравнивающегося участка. |
 |
| Рис.9. Выравнивания структурных гомологов структуры 1J0X (JalView, раскраска Clustalx по консервативности 30%). |
Совмещение структур альфа и бета доменов константной части T-клеточного рецептора
Была выбрана структура 1OGA: альфа-цепь region d:118-202, бета-цепь region e:119-245.
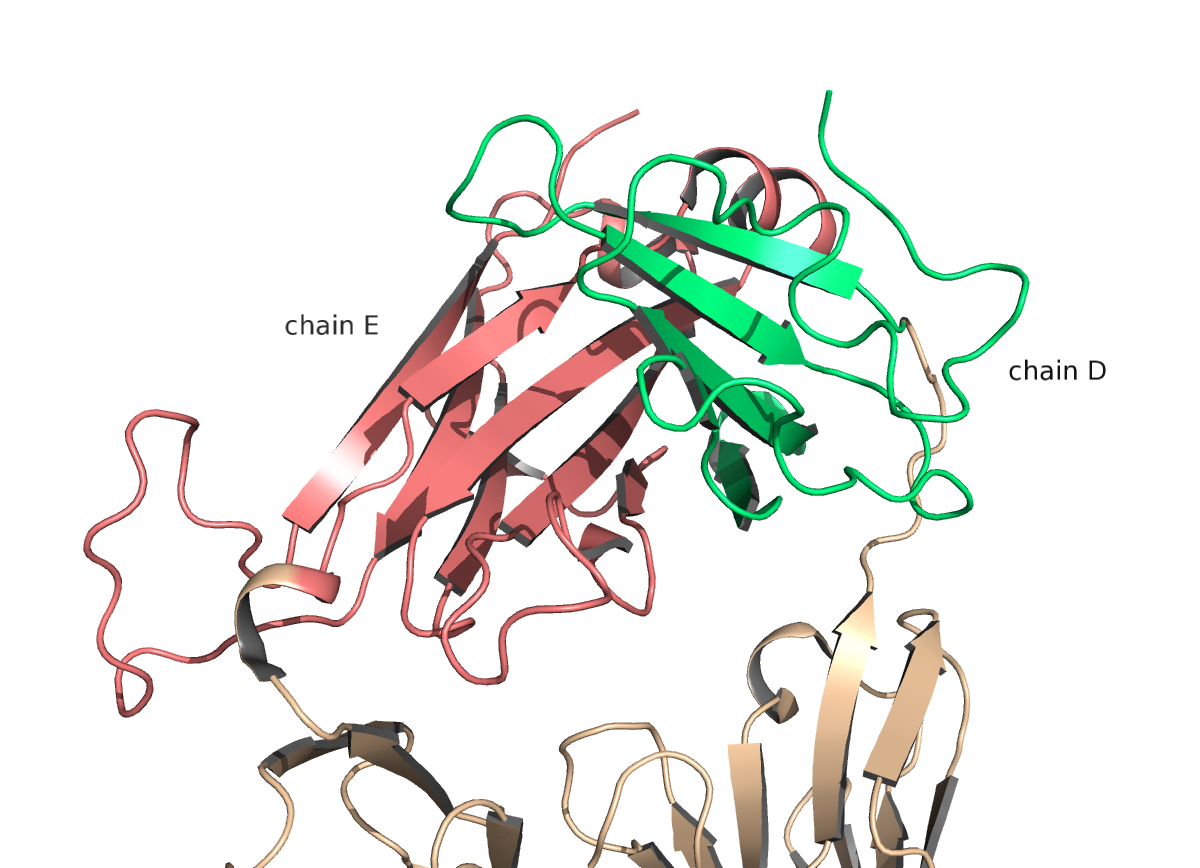 |
| Рис.10. Альфа и бета домены структуры 1OGA. |
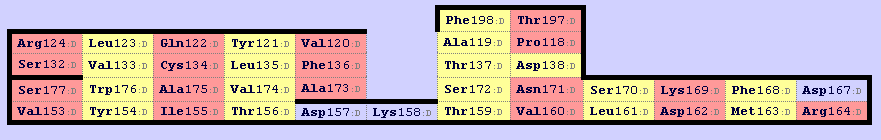 |
| Рис.11. SheeP карта бета-листа для альфа-цепочки. |
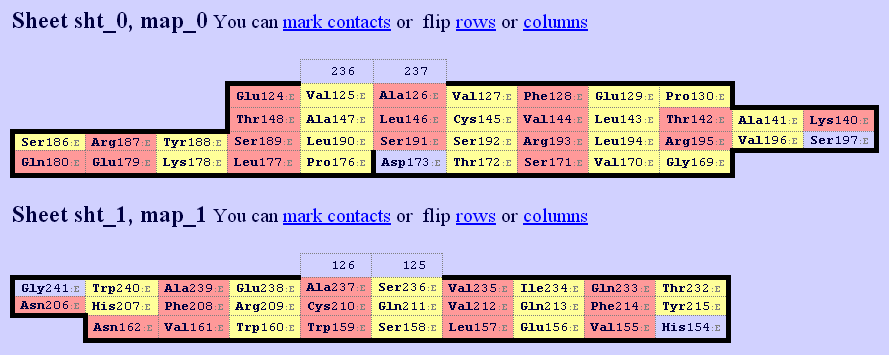 |
| Рис.12. SheeP карта бета-листа для бета-цепочки. |
Два консервативных цистеина Cys134:D (альфа-домен) и Cys145:E (бета-домен) отмечены желтым. Выравнивание структур бета-листа альфа-домена и бета-листов бета-домена приведено на рисунке 13. Для выравнивания были взяти девять СА атомов вокруг цистеина (включая его СА атом, остаток отмечен желтым). Для удобства сравнения - только бета-тяжи, так как петли все равно различаются и являются нерегулярными и более подвижными элементами структуры.
 |
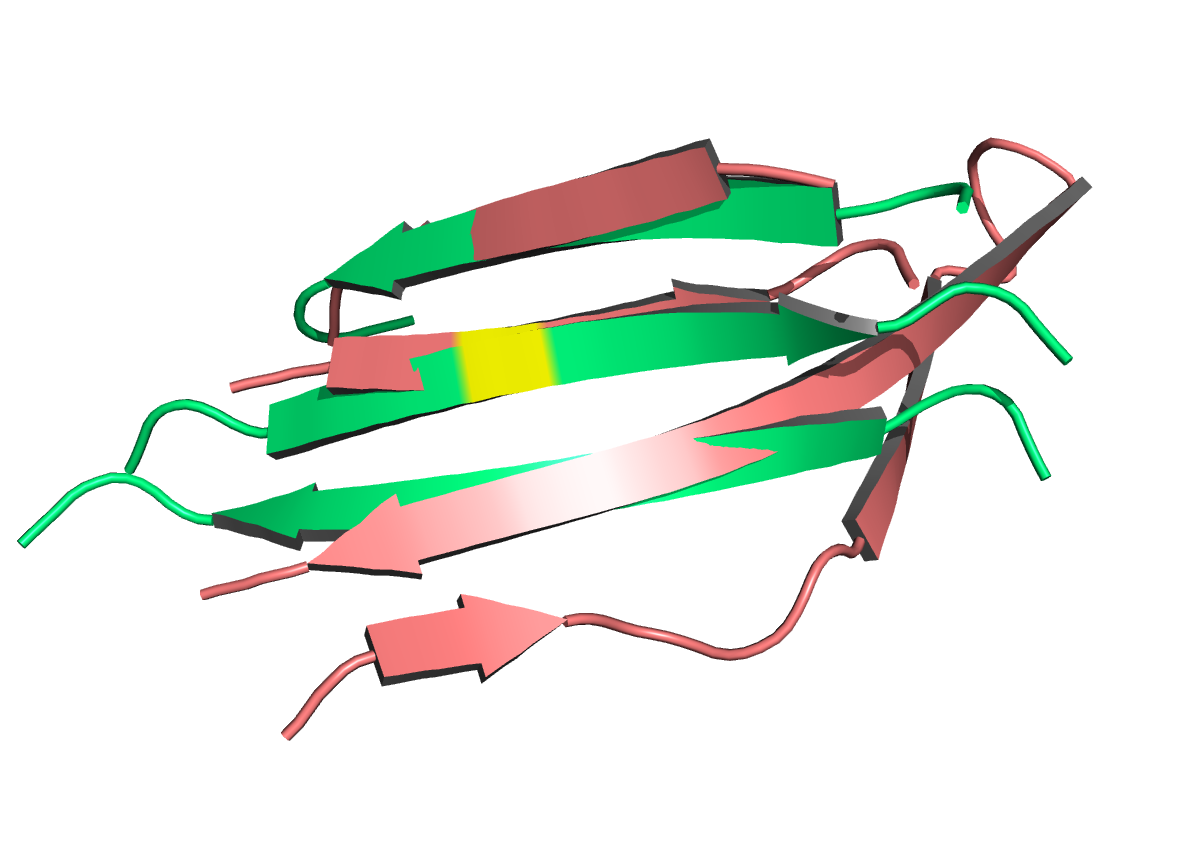 |
Рис.13. Выравнивание структур бета-листа альфа-домена и первого (ближе к альфа-домену) бета-листа бета-домена.
pair_fit alpha_list///121-123+133-135+174-176/CA,
beta_1///126-128+144-146+191-193/CA |
Рис.13. Выравнивание структур бета-листа альфа-домена и второго (дальше от альфа-домена) бета-листа бета-домена.
pair_fit alpha_list///121-123+133-135+174-176/CA,
beta_2///236-238+209-211+158-160/CA |
RMSD первого наложения 0,485, второго 0,548, хотя визуально я бы сказала, что со вторым листом бета-домена выравнивается лучше. Я считаю, что сходство топологий наблюдается во втором случае, так как качество наложения не сильно хуже, но зато количество и направление бета-тяжей совпадает.
Скрипт для получения изображений.
d3. Нахождение гидрофобных кластеров
Найдем гидрофобные кластеры в структуре соматической кроличьей ГАФД ( PDB-код - 1J0X ). Для этого будем использовать Clud.
Parameters:
- Atom list: strict
- Model number (for NMR): 1
- All chains selected
- Distance threshold: 4.5 Å (при большем расстоянии туда сможет поместиться молекула воды)
- Cluster size threshold: 5;
Output files:
При запуске алгоритма с такими параметрами всего нашлось 76 кластеров, а больше заданного размера - 42. Два самый больших (Рис.14) представляют собой как бы объединение цепей парами, включая из NAD+-связывающие домены (это укладка Россмана => там есть гидрофобные контакты между тяжами и спиралями). Так же отдельны были найдены функциональные домены для всех цепей - каталитический (Рис.15) и NAD+-связывающий домены, что несет уже больше биологического смысла (Рис.16). Так же обнаружились гидрофобные контакты между цепями (Рис.17-21).
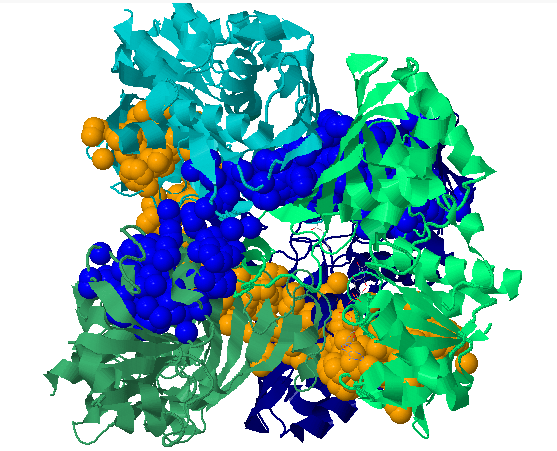 |
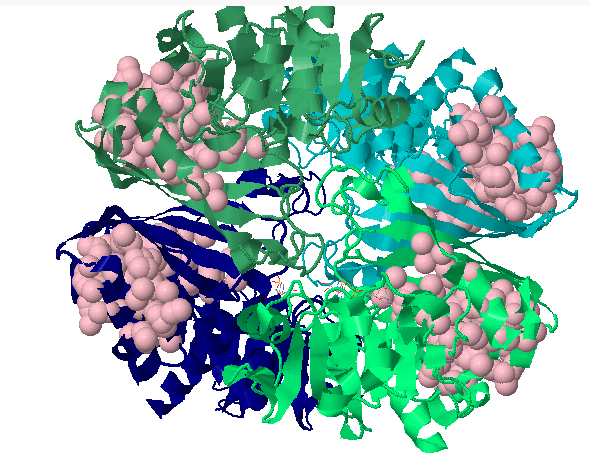 |
| Рис.14. Два самый больших найденных гидрофобных кластера (напоминают interlock по пространственному расположению =)). |
Рис.15. C-концевой домен каталитический домены |
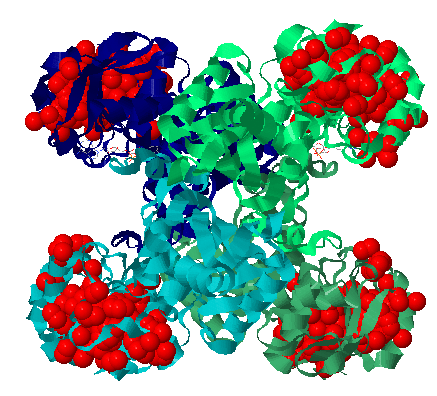 |
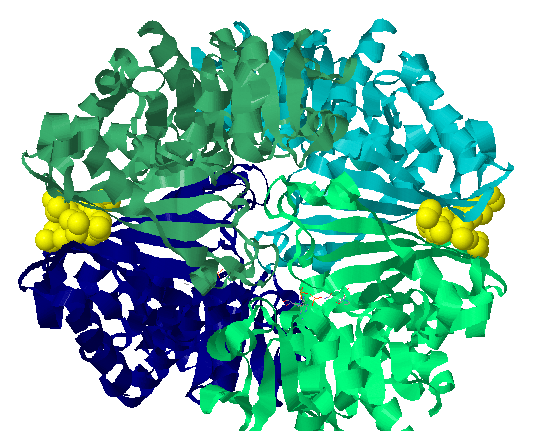 |
| Рис.16. NAD+-связывающие домены |
Рис.17. Взаимодействия между цепями Q-R и P-Q. |
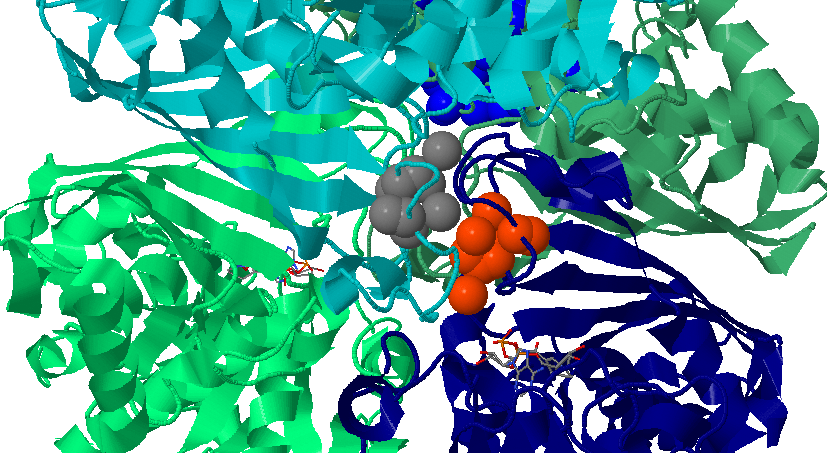 |
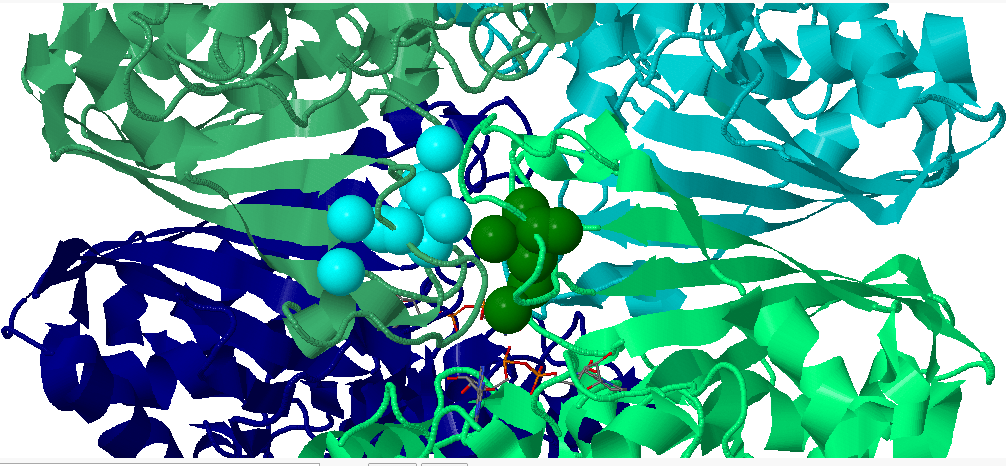 |
| Рис.18. Взаимодействия между цепями O-R. |
Рис.19. Взаимодействия между цепями O-Q. |
 |
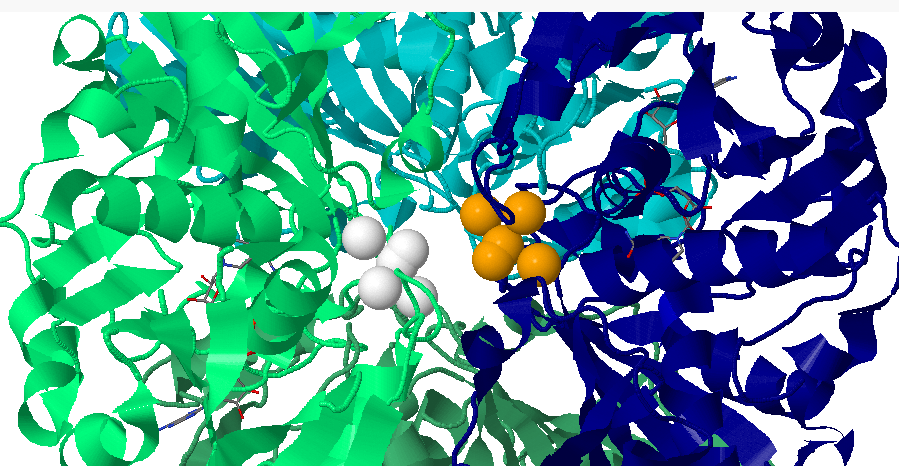 |
| Рис.20. Взаимодействия между цепями P-O. |
Рис.21. Взаимодействия между цепями P-R. |
d4. Построение поверхности, раскраска участка поверхности: PyMol
Для комплекса димера пуринового репрессора с ДНК (PDB-код 1QP7) с помощью средств PyMol были созданы изображения:
- а) поверхности контакта мономера белка с симметричным мономером на фоне остовной (ribbon) модели мономера;
- б) поверхности контакта димера белков с двойной спиралью ДНК на фоне остовной модели части белка, вовлечённой в контакт;
- в) поверхности контакта ДНК с димером белков на фоне проволочной (sticks) модели двойной спирали.
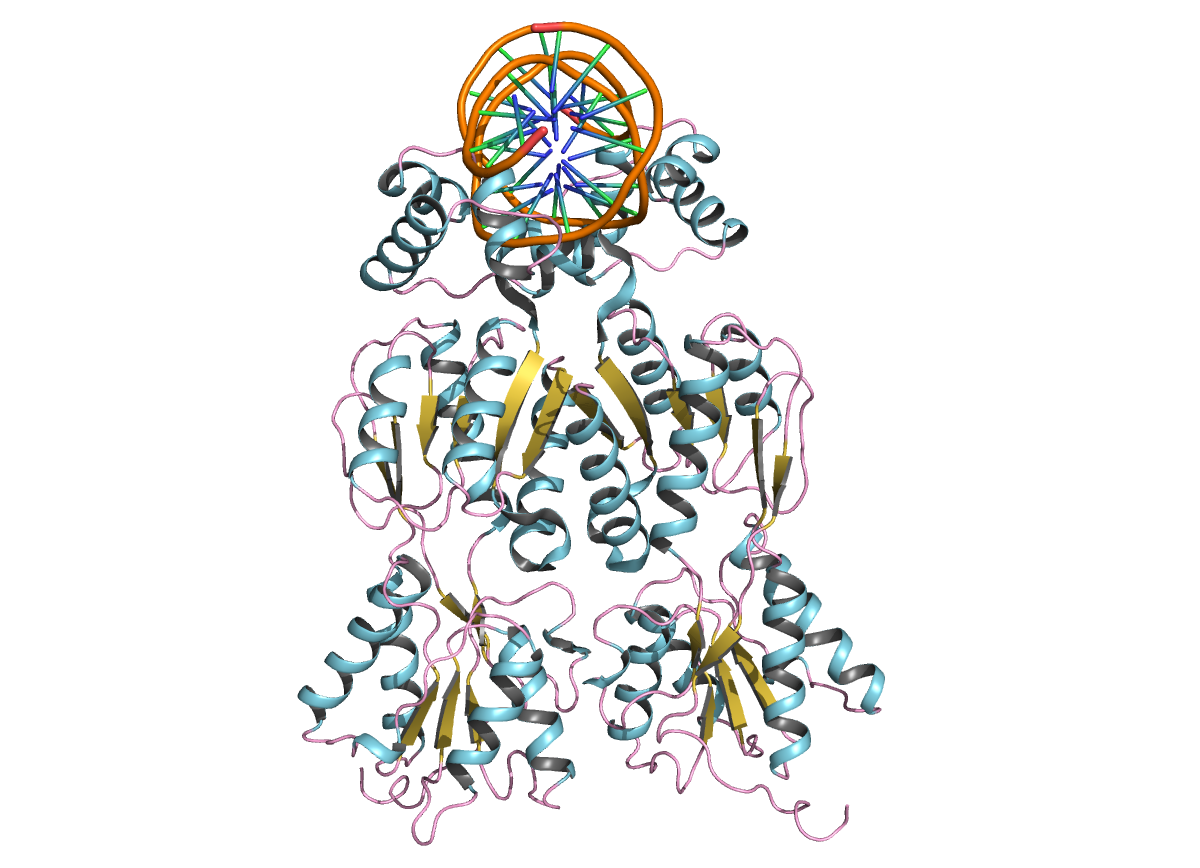 |
| Рис.22. Изображение димера белка в комплексе с ДНК. |
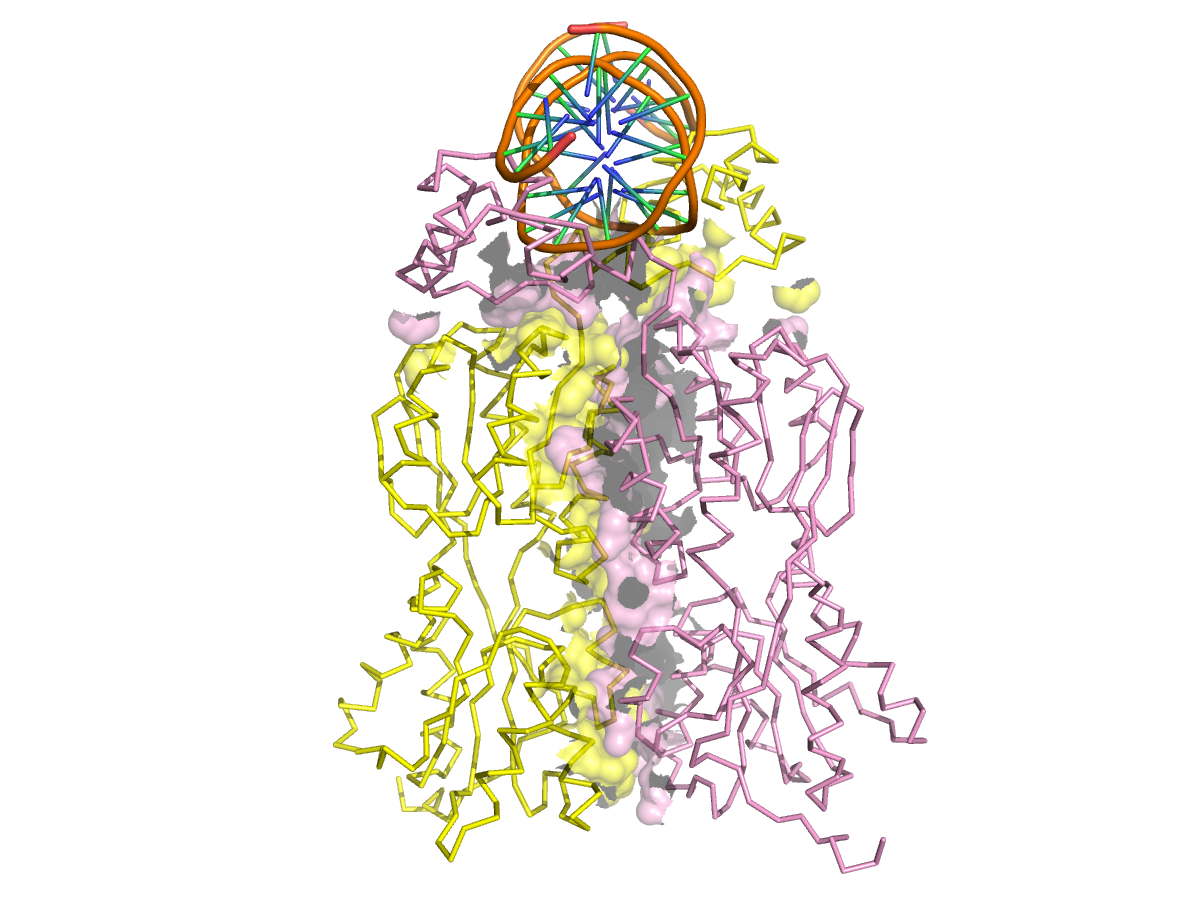 |
| Рис.23. Изображение поверхности контакта между мономерами белка. Контакт определялся расстоянием меньше или равном 5.0 ангстрем. Контакты с ДНК не учитывались. |
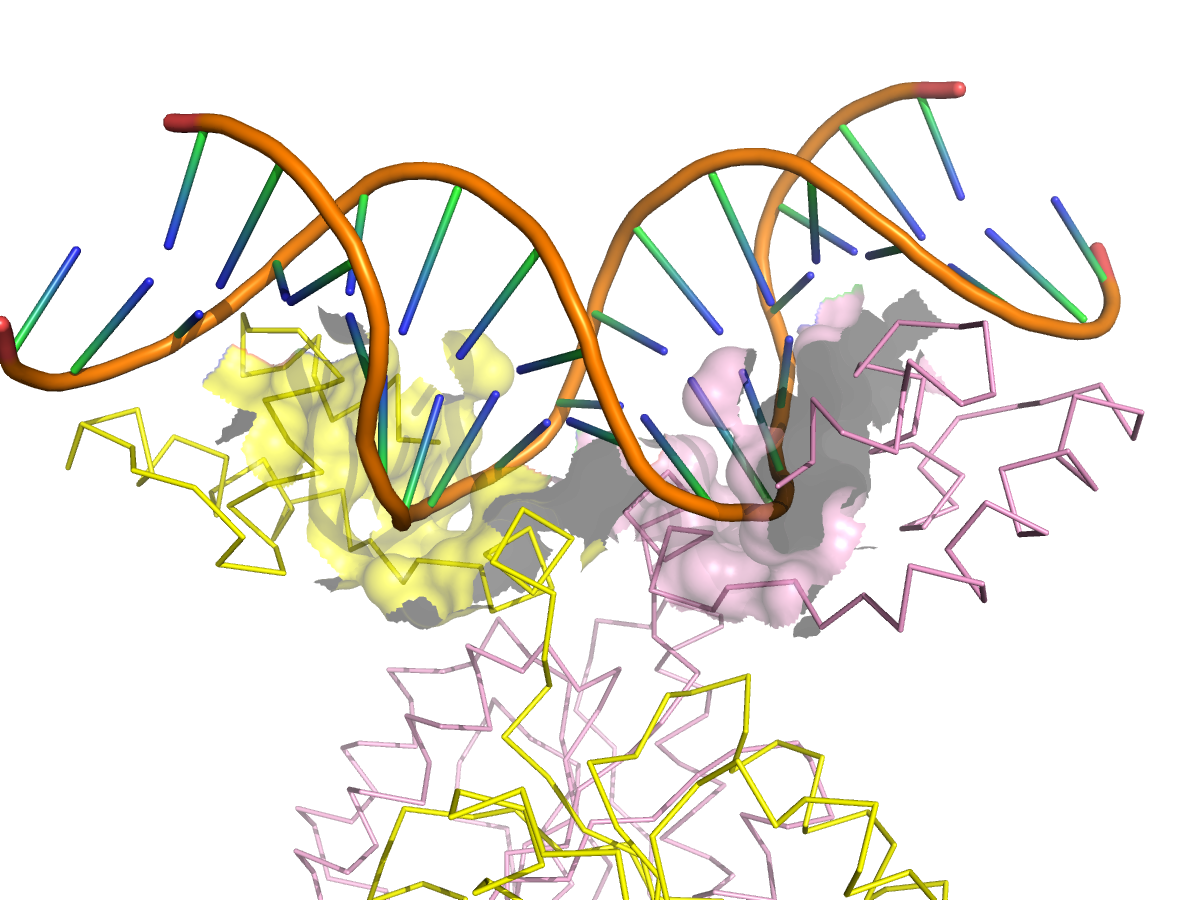 |
| Рис.24. Изображение поверхности контакта димера белков с двойной спиралью ДНК на фоне остовной модели части белка, вовлечённой в контакт. |
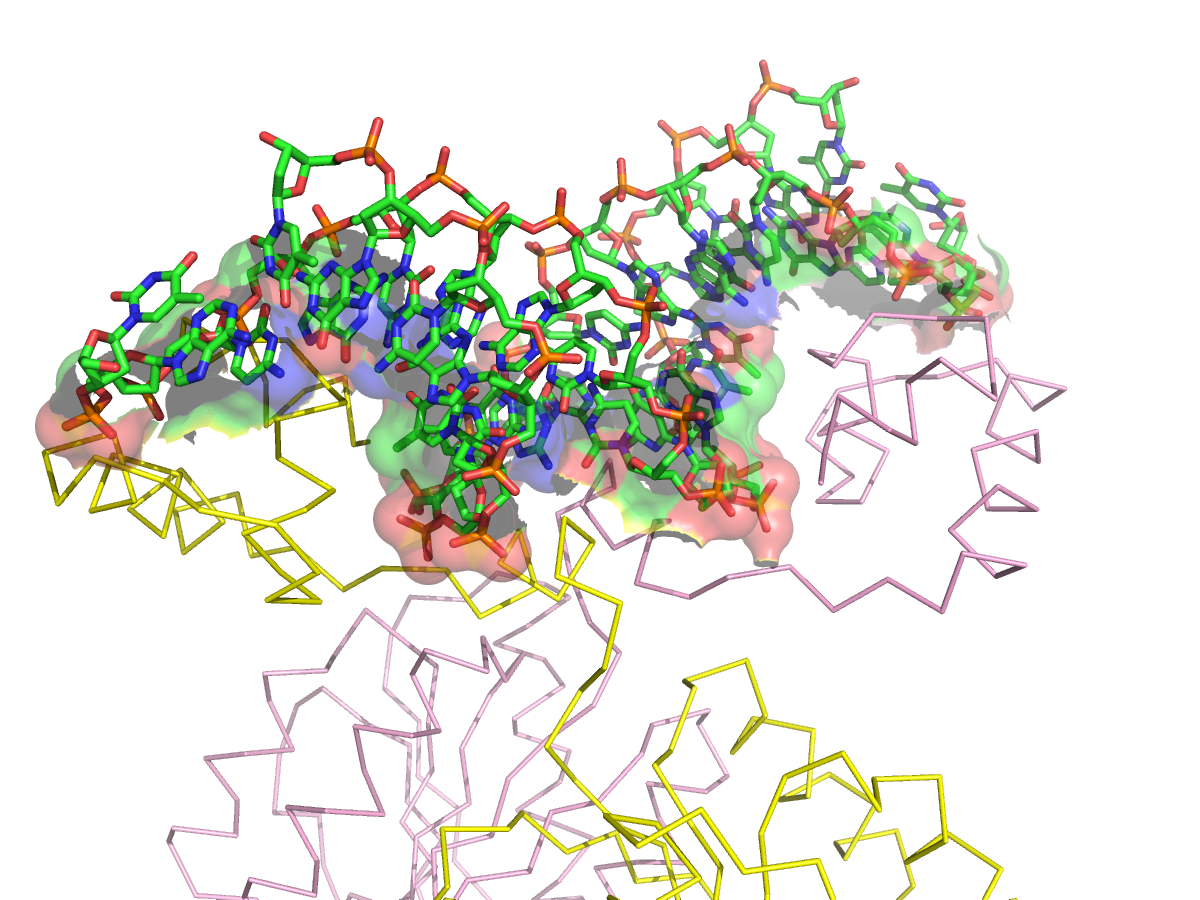 |
| Рис.25. Изображение поверхности контакта ДНК с димером белков на фоне проволочной (sticks) модели двойной спирали. |
С помощью сервиса CluD, были определены гидрофобные кластеры объёмом не менее 10 атомов на интерфейсе мономеров белка в том же комплексе (Рис. 26) - их получилось 4.
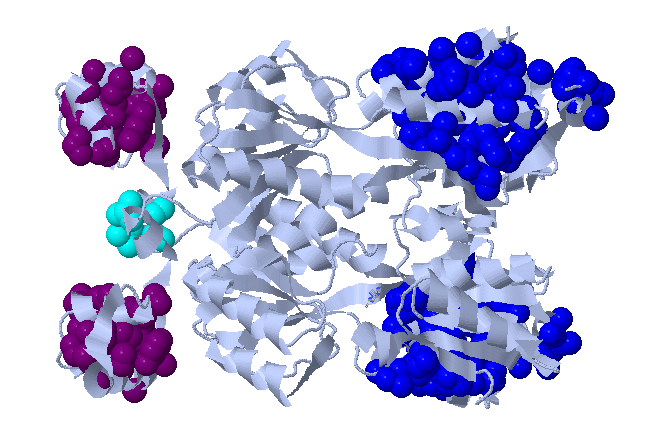 |
| Рис.26. Гидрофобные кластеры между мономерами, найденные CluD. |
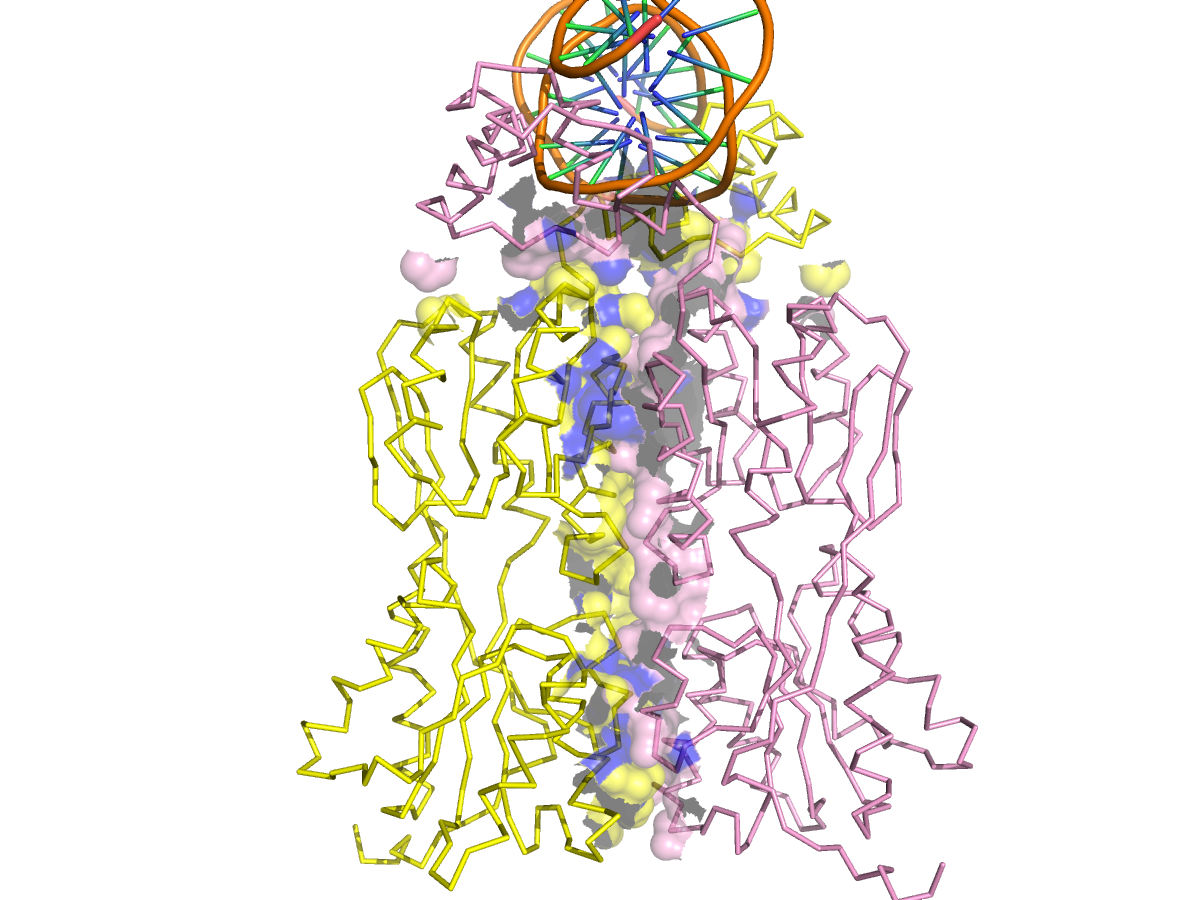 |
| Рис.27. Изображение поверхностей атомов мономера белка, образующих гидрофобные контакты с другим мономером. Атомы, входящие в гидрофобные кластеры, найденные CluD, окрашены синим. |
Скрипт для получения изображений.
d5. Сравнение доменов SCOP и Pfam
Для человеческого кальмодулина (Uniprot ID CALM_HUMAN, PDB структура 1CLL) найдем границы доменов в SCOP и Pfam. Поиск человеческого кальмодулина по PDB ID по базе данных Pfam обнаружил наличие двух доменов EF-hand (координаты 7-73 и 85-146). Поиск по SCOP - ничего адекватного относительно координат не дал, однако в классификации указано, что там две пары кальций-связывающих доменов EF-hands, а для всех других записей определил координаты как на весь белок целиком почти от самого начала цепи. Поэтому я для точности посмотрела еще описание доменов в Uniprot. Благодаря разметке Uniprot можно понять, что коротелькийбета-тяж+петля+кусок альфа-спирали составляют центр связывания кальция, и две пары кальций-связывающих доменов (домены идут прямо один за другим) действительно разделены альфа-спиральным шарниром, как и положено.
 |
 |
| Рис.28. Домены Pfam. |
Рис.29. Домены SCOP. |
 |
| Рис.30. Классификация SCOP. |
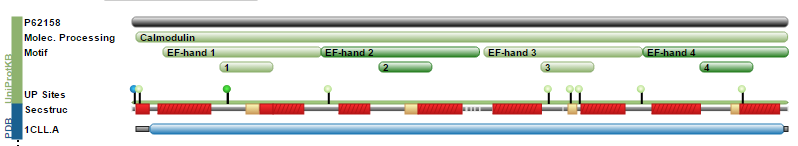 |
| Рис.31. Домены Uniprot. |
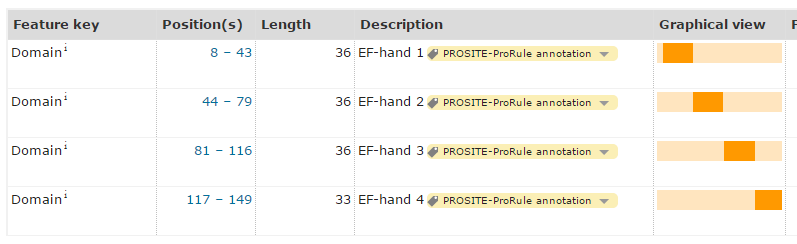 |
| Рис.32. Домены Uniprot. |
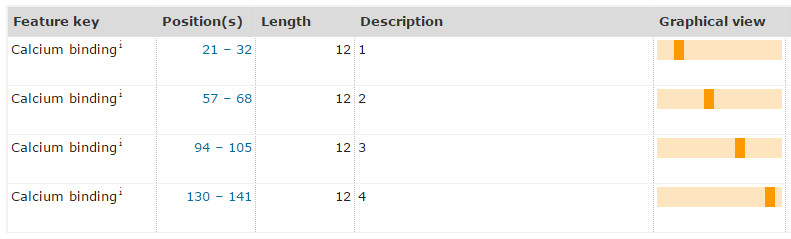 |
| Рис.33. Участки связывания кальция Uniprot. |
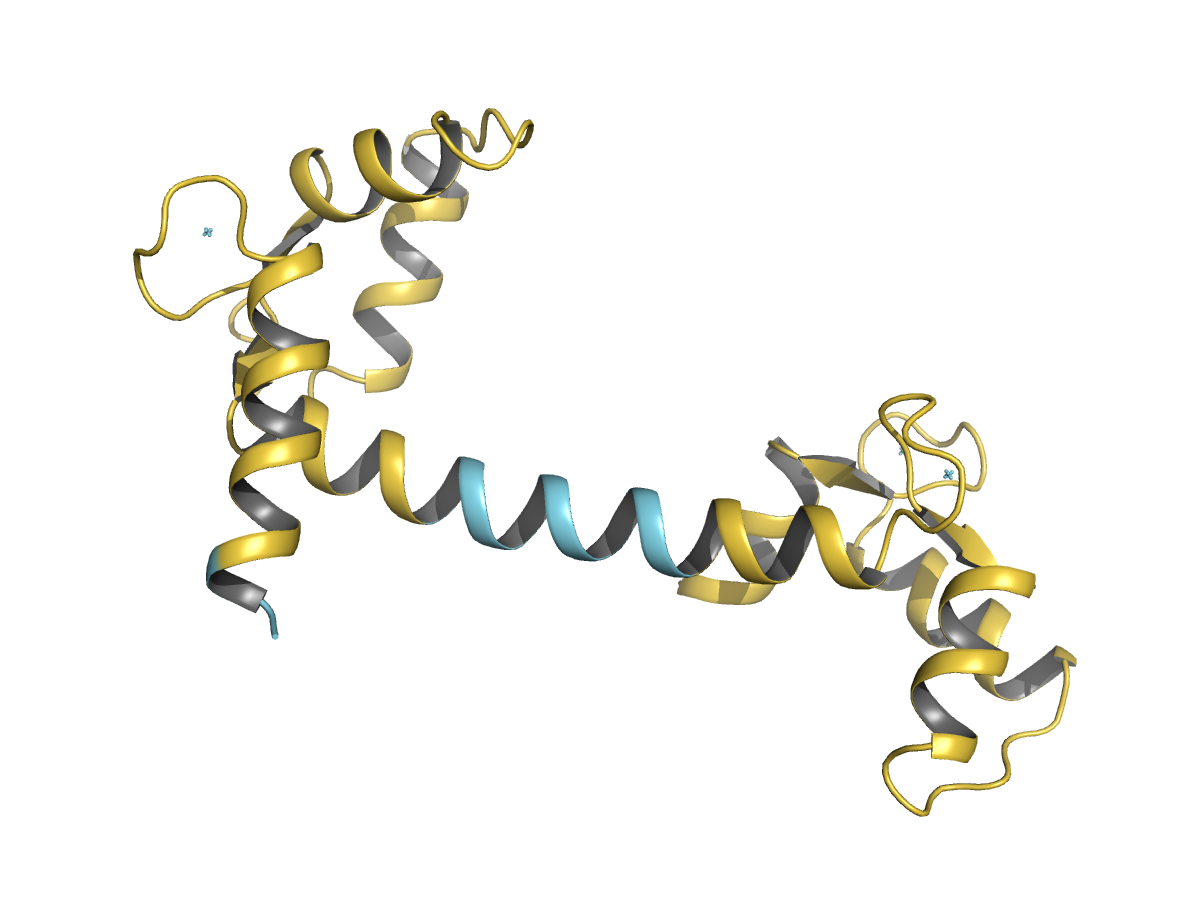 |
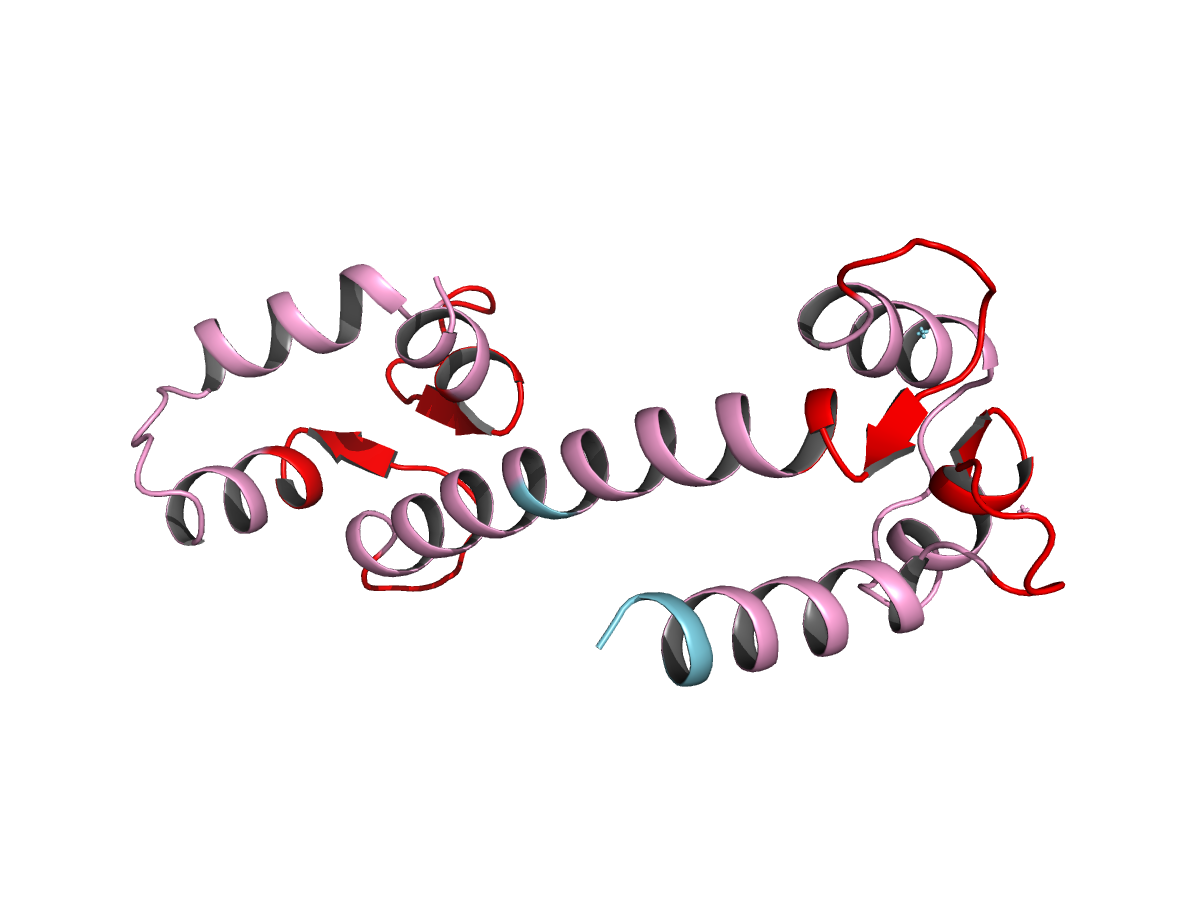
|
| Рис.34. Домены Pfam (желтым, 7-73, 85-146). |
Рис.35. Домены Uniprot (розовым, 8-43, 44-79, 81-116, 117-149; красным - кальций-связывающие центры). |
Скрипт для получения изображений.
|