Abinitio вычисления для нафталена и азулена
Суть задания состоит в поэтапном освоении возможностей GAMESS как стандартного квантово-химического пакета. В качестве примера найдем оптимальную геометрию для нафталена и азулена и рассчитаем теплоты образования этих молекул разными подходами квантовой механики.
Рабочая директория /home/students/y11/janemoiseeva/Term8/Pr3
Построение структур нафталена и азулена
SMILES этих молекул:
Azulene : C1=CC=C2C=CC=C2C=C1 Napthalene: c1ccc2ccccc2c1
Создадим файлы .smi со описанием структур и на основе этих описаний c помощью программы из openbabel построим 3D структуры:
echo "C1=CC=C2C=CC=C2C=C1 azulene" > azulene.smi obgen azulene.smi > azulene.mol echo "c1ccc2ccccc2c1 naphthalene" > naphthalene.smi obgen naphthalene.smi > naphthalene.mol babel -imol azulene.mol -omop azulene.mop -xk "PM6" babel -imol naphthalene.mol -omop naphthalene.mop -xk "PM6" MOPAC2009.exe azulene.mop MOPAC2009.exe naphthalene.mop babel -imopout azulene.out -opdb azulene.pdb babel -imopout naphthalene.out -opdb naphthalene.pdb
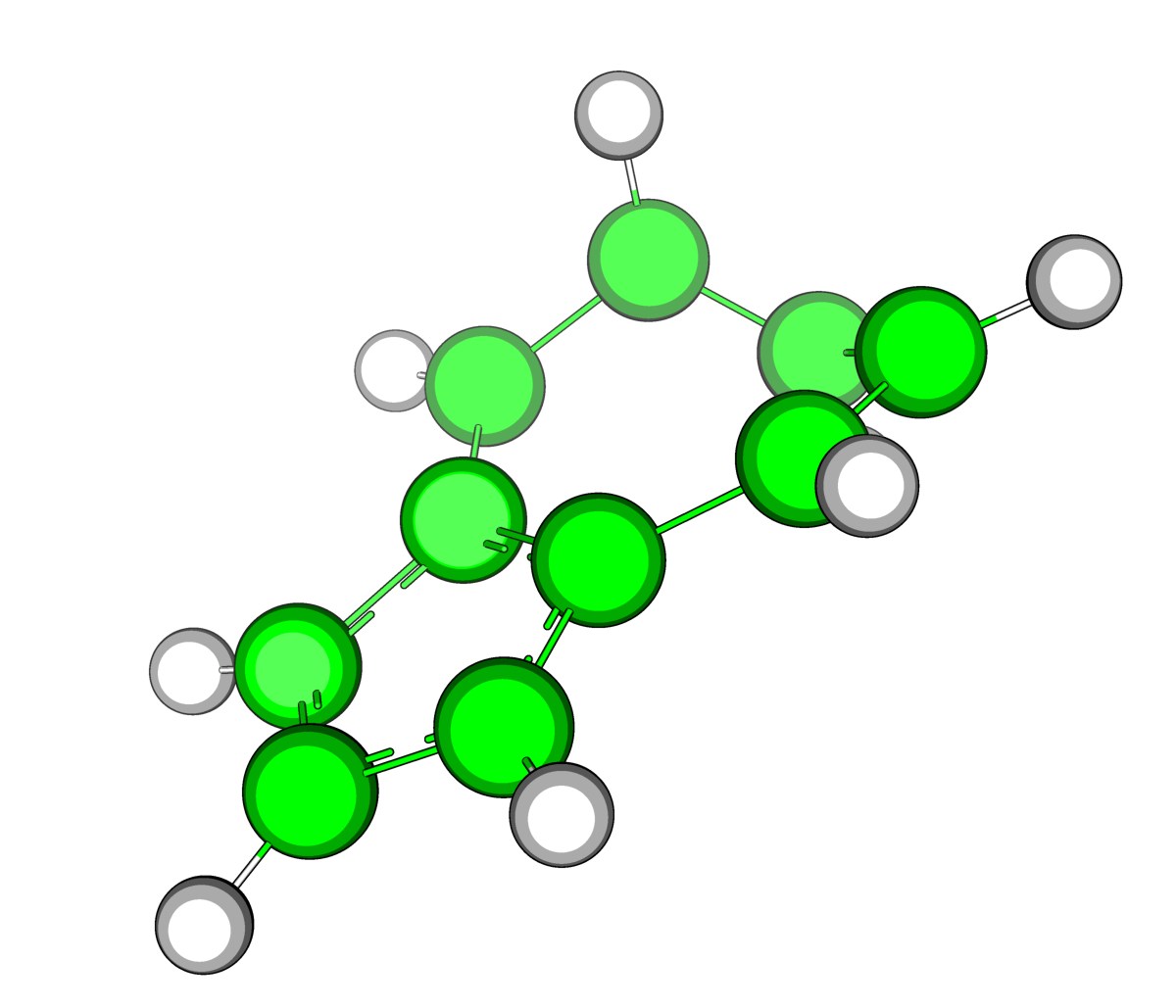 |
 |
| Рис.1.Оптимизированные в Mopac структуры азулена(слева) и нафталена (справа) | |
С плоской геометрией молекулы нафталена проблем нет - она как и ожидась такова. Однако, молекула азулена не плоская. Чтобы это исправить, попробуем использовать силовое поле UFF (c MMFF94 не оплучилось) в obgen.
obgen azulene.smi -ff UFF > azulene_UFF.mol babel -imol azulene_UFF.mol -omop azulene_UFF.mop -xk "PM6" MOPAC2009.exe azulene_UFF.mop babel -imopout azulene_UFF.out -opdb azulene_UFF.pdb
 |
| Рис.2. Уплощенная при помощи силового поля UFF (параметр obgen) структура азулена |
Подготовка входных файлов для оптимизации геометрии средствами GAMESS
Для этого переформатируем координаты, записанные в файлах naphthalene.out и azulene_UFF.out в gamin формат и отредактируем заголовок:
babel -imopout azulene_UFF.out -ogamin azulene.gamin babel -imopout naphthalene.out -ogamin naphthalene.gamin HEADER=" \$CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=OPTIMIZE \$END\n\ \$BASIS GBASIS=N31 NGAUSS=6 \$end\n\ \$system mwords=2 \$end\n\ \$DATA" echo -en " " > azu_opt.inp echo -e $HEADER >> azu_opt.inp tail -n+4 azulene.gamin >> azu_opt.inp echo -en " " >> nap_opt.inp echo -e $HEADER > nap_opt.inp tail -n+4 naphthalene.gamin >> nap_opt.inp
Из заголовка понятно, что оптимизацию будем проводить с базисом 6-31G - Pople's N-31G split valence basis set, где N - число гауссианов(у нас 6). При использовании такого базиса орбитали остова представлены шестью гауссианами, а валентные орбитали представлены двумя составляющими — из трёх гауссианов и из одного гауссиана.
Оптимизация геометрии средствами GAMESS
Проведём оптимизацию геометрии обеих молекул средствами GAMESS:
gms nap_opt.inp 1 >& nap_opt.log gms azu_opt.inp 1 >& azu_opt.log
И отобразим полученные структуры в PyMol:
babel -igamout azu_opt.log -opdb azu_gamess.pdb babel -igamout nap_opt.log -opdb nap_gamess.pdb
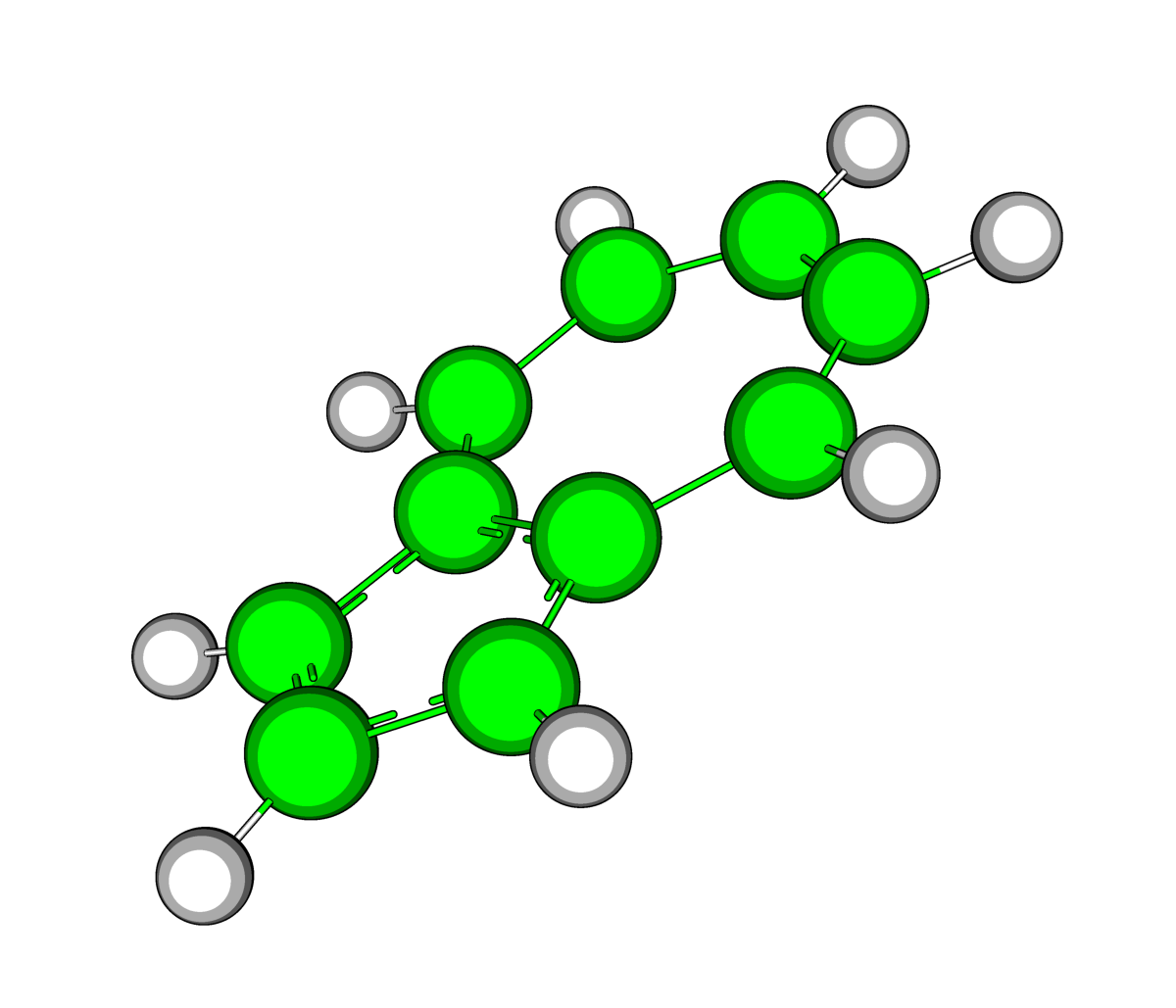
 |
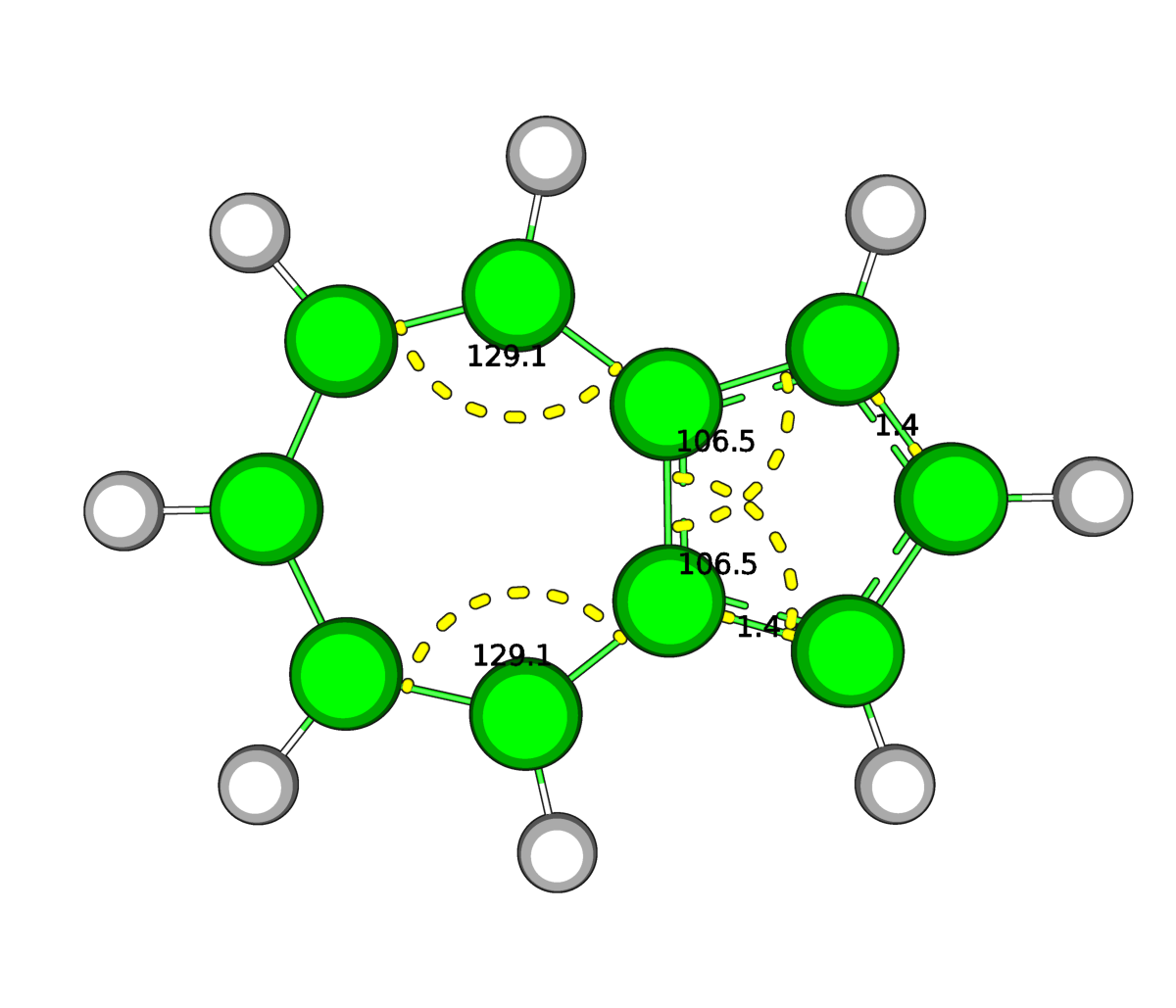 |
| Рис.3.Оптимизированная в Mopac (слева) и средствами GAMESS(справа) структура азулена | |
В результате оптимизации в структуре азулена незначительно изменились длины двух связей между углеродами пятичленного кольца (показаны на рисунке), а также почти все углы на 0,5-1 градус (показаны некоторые).
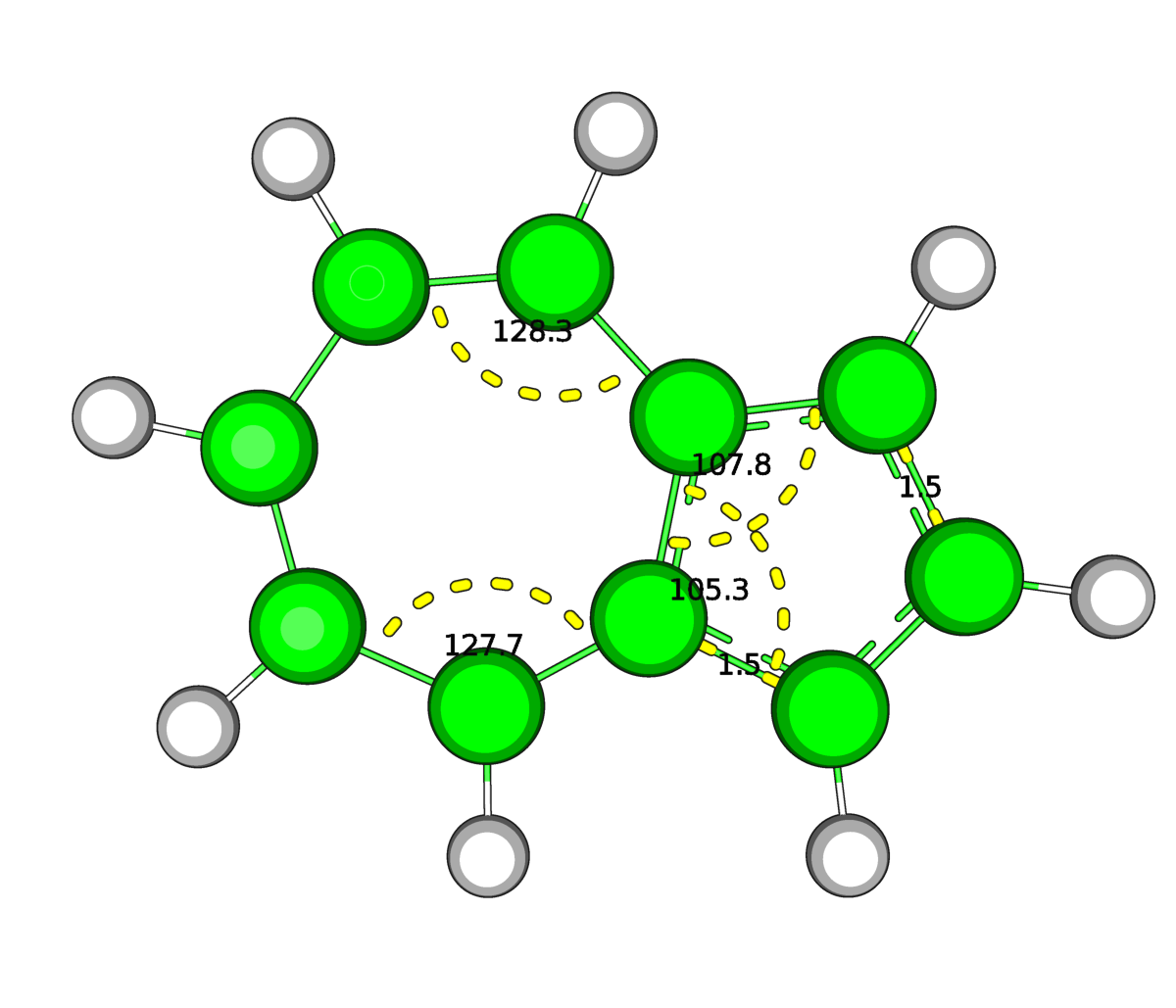
 |
 |
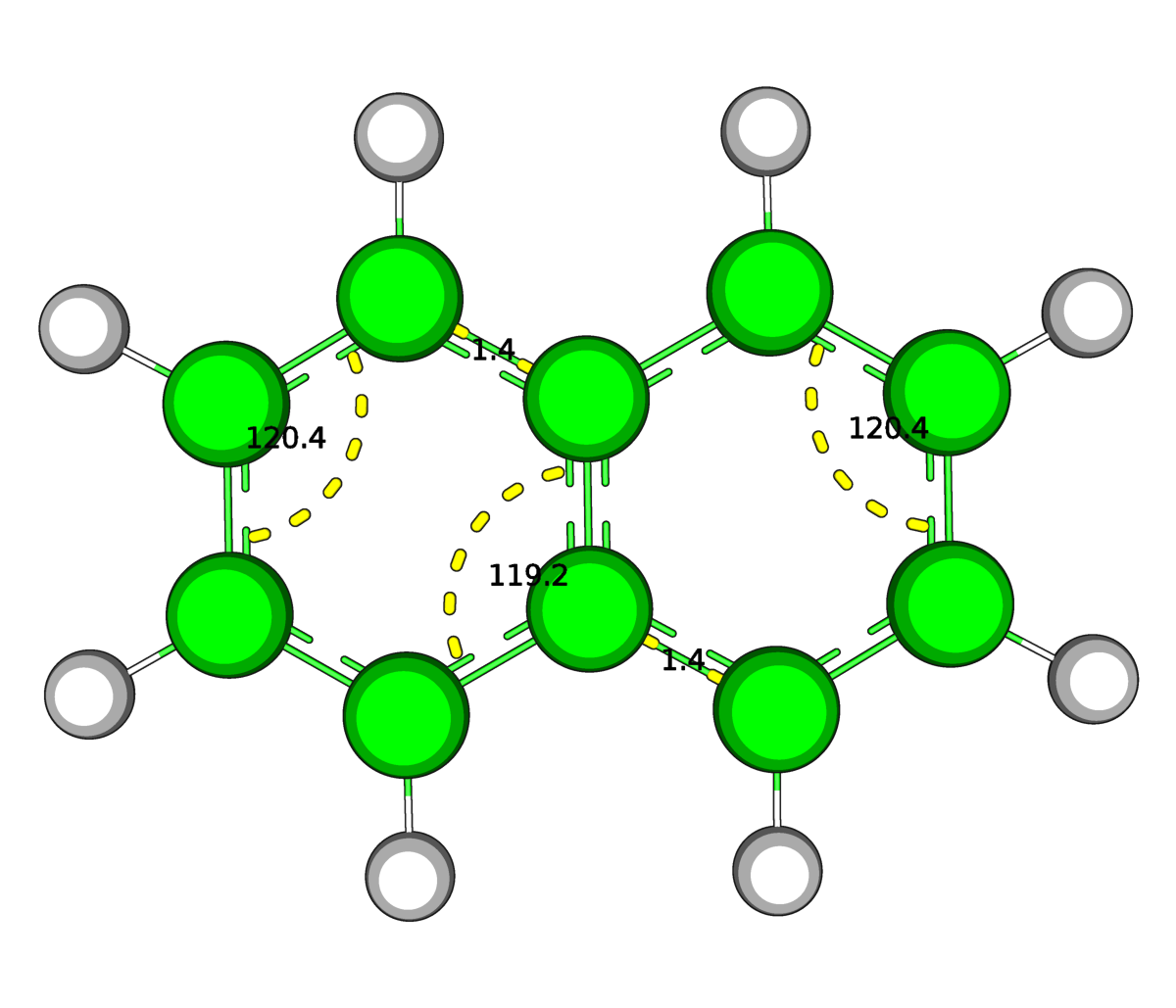
| Рис.4.Оптимизированная в Mopac (слева) и средствами GAMESS(справа) структура нафталена | |
В структуре нафталена после выполнения оптимизации изменений не обнаружилось (на рисунке приведены некоторые углы и связи в качестве примера). Обе структуры как были, так и остались плоскими.
На основе полученных координат составим новые входные файлы для расчёта энергии: для расчёта методом Хартри-Фока (Hartree-Fock) и для использования теории функционала плотности (DFT - Density functional theory).
Расчёт методом Хартри-Фока
Подготовим входные файлы для расчёта по Хартри-Фоку:
babel -igamout azu_opt.log -ogamin azu_gamout.inp babel -igamout nap_opt.log -ogamin nap_gamout.inp HEADER="\$CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=ENERGY \$END\n\ \$BASIS GBASIS=N31 NGAUSS=6\n\ POLAR=POPN31 NDFUNC=1 \$END\n\ \$GUESS GUESS=HUCKEL \$END\n\ \$system mwords=2 \$end\n\ \$DATA" echo -en " " > azu_HF.inp echo -e $HEADER >> azu_HF.inp tail -n+4 azu_gamout.inp >> azu_HF.inp echo -en " " > nap_HF.inp echo -e $HEADER >> nap_HF.inp tail -n+4 nap_gamout.inp >> nap_HF.inpС помощью GAMESS рассчитаем энергию:
gms nap_HF.inp 1 >& nap_HF.log gms azu_HF.inp 1 >& azu_HF.log grep "TOTAL ENERGY =" azu_HF.log nap_HF.logПолучили:
azu_HF.log: TOTAL ENERGY = -383.2825020374 nap_HF.log: TOTAL ENERGY = -383.3523603875
Расчёт с использованием теории функционала плотности
Так же подготовим входные файлы для расчёта:HEADER="\$CONTRL COORD=CART UNITS=ANGS dfttyp=b3lyp RUNTYP=ENERGY \$END\n\ \$BASIS GBASIS=N31 NGAUSS=6\n\ POLAR=POPN31 NDFUNC=1 \$END\n\ \$GUESS GUESS=HUCKEL \$END\n\ \$system mwords=2 \$end\n\ \$DATA" echo -en " " > azu_DFT.inp echo -e $HEADER >> azu_DFT.inp tail -n+4 azu_gamout.inp >> azu_DFT.inp echo -en " " > nap_DFT.inp echo -e $HEADER >> nap_DFT.inp tail -n+4 nap_gamout.inp >> nap_DFT.inpИ рассчитаем энергию:
gms nap_DFT.inp 1 >& nap_DFT.log gms azu_DFT.inp 1 >& azu_DFT.log grep "TOTAL ENERGY =" azu_DFT.log nap_DFT.logПолучили:
azu_DFT.log: TOTAL ENERGY = -385.5857377223 nap_DFT.log: TOTAL ENERGY = -385.6417198702
Сравнение методов подсчета теплот образования
| Вещество | ENaphthalene | EAzulene | ΔE, Hartree* | ΔE, kCal/mol* |
|---|---|---|---|---|
| Хартри-Фок | -383.3523603875 | -383.2825020374 | 0,06985835 | 43,83681327 |
| DFT | -385.6417198702 | -385.5857377223 | 0,055982148 | 35,12935763 |
Из эксперимента известно, что энергия изомеризации нафталина в азулен составляет 35.3±2.2 kCal/mol, что численно ближе к результатам подсчета по DFT.
Вся работа в PyMol была записана в скрипт: script_1.pml, а рабочая директория, где лежат все файлы расчетов - /home/students/y11/janemoiseeva/Term8/Pr3.