Гомологичное моделирование комплекса белка с лигандом
Для работы был взят лизоцим бактериофага PS119 -
LYS_BPPS1.
Программе MODELLER для моделирования структуры белков, в качестве входных данных нужны:
- управляющий скрипт
- файл pdb со структурой-образцом
- файл выравнивания с дополнительной информацией.
Построение выравнивания
Построим выравнивание последовательности из структуры PDB ID: 1lmp и белка LYS_BPPS1 при помощи программы ClustalW и сохраним полученное выравнивание в формате PIR.
Модификация выравнивания
Последовательность в файле выравнивания переименовали как в примере: P1;seq и P1;1lmp. После имени последовательности моделируемого белка добавим строчку (описывает входные параметры последовательности для modeller):
sequence:ХХХХХ::::::: 0.00: 0.00После имени последовательности белка-образца добавим строчку (описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи...):
structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00В конце каждой последовательности добавим символы:
/. "/" конец цепи белка "." один лигандПолучили файл - aligned.pir.
Модификация файла со структурой
Удалим всю воду из структуры (в текстовом редакторе). Затем всем атомам лиганда присвойте один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицируем имена атомов каждого остатка, добавив в конец буквы A, B, C. Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. Сохраним новый файл как 1lmp_now.ent
Создание управляющего скрипта lys_bpps1.py
Дана заготовка скрипта. Необходимо ее модифицировать - отредактировать строчки, в которых указано, какие водородные связи белка с лигандом должны быть в будущей модели. Для этого нужно определим, какие водородные связи между белком и лигандом присутствуют в образце (Рис.1). В пробном белке 167 а.к., значит номер "остатка" лиганда 168, а номера остатков в белке надо определяем согласно выравниванию. Так же "цепь" лиганда будем обозначать "В", т.к. в выравнивании конец цепи белка обозначался "/", т.е. лиганд - новая цепь.
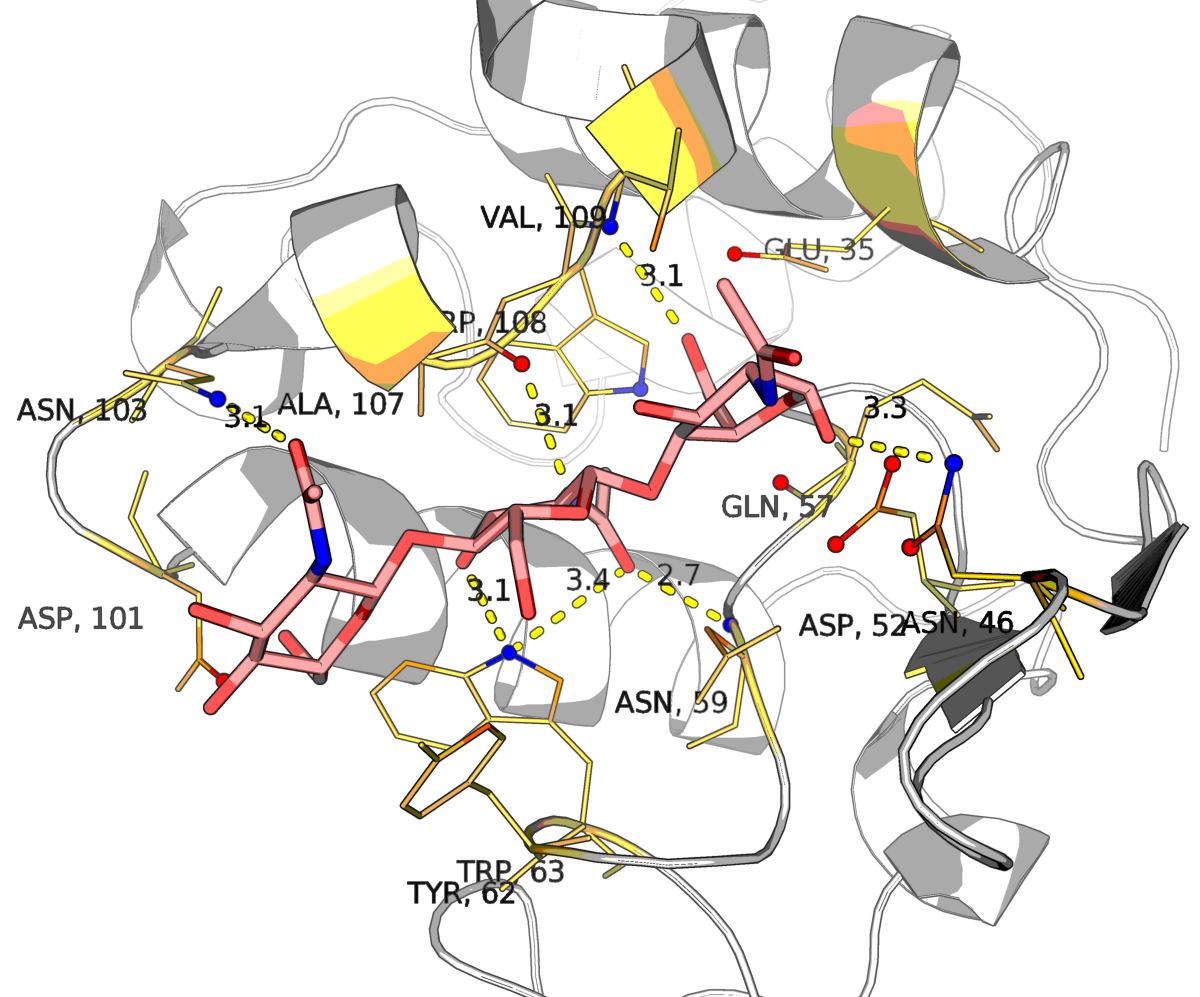 |
| Рис.1. Водородные связи между белком и лигандом в структуре 1lmp |
| Было | Стало |
| N:109:A - O6:130:A | N:105:A - O6C:168:B |
| O:107:A - N2:131:A | O:103:A - N2B:168:B |
| N:59:A - O7:131:A | N:54:A - O7B:168:B |
mod9v7 lys_bpps1 &После запуска скрипта мы получили пять моделей вирусного белка с лигандом:
Сравнение полученных моделей
Все модели в участке контакта с лигандом (да и везде вообще) очень похожи друг на друга, поэтому визульно оценить их качество нельзя. Есть некоторые расхождения наблюдаются в петлях и N-конце, который никак не структурирован, так как в структуре образца его не было.
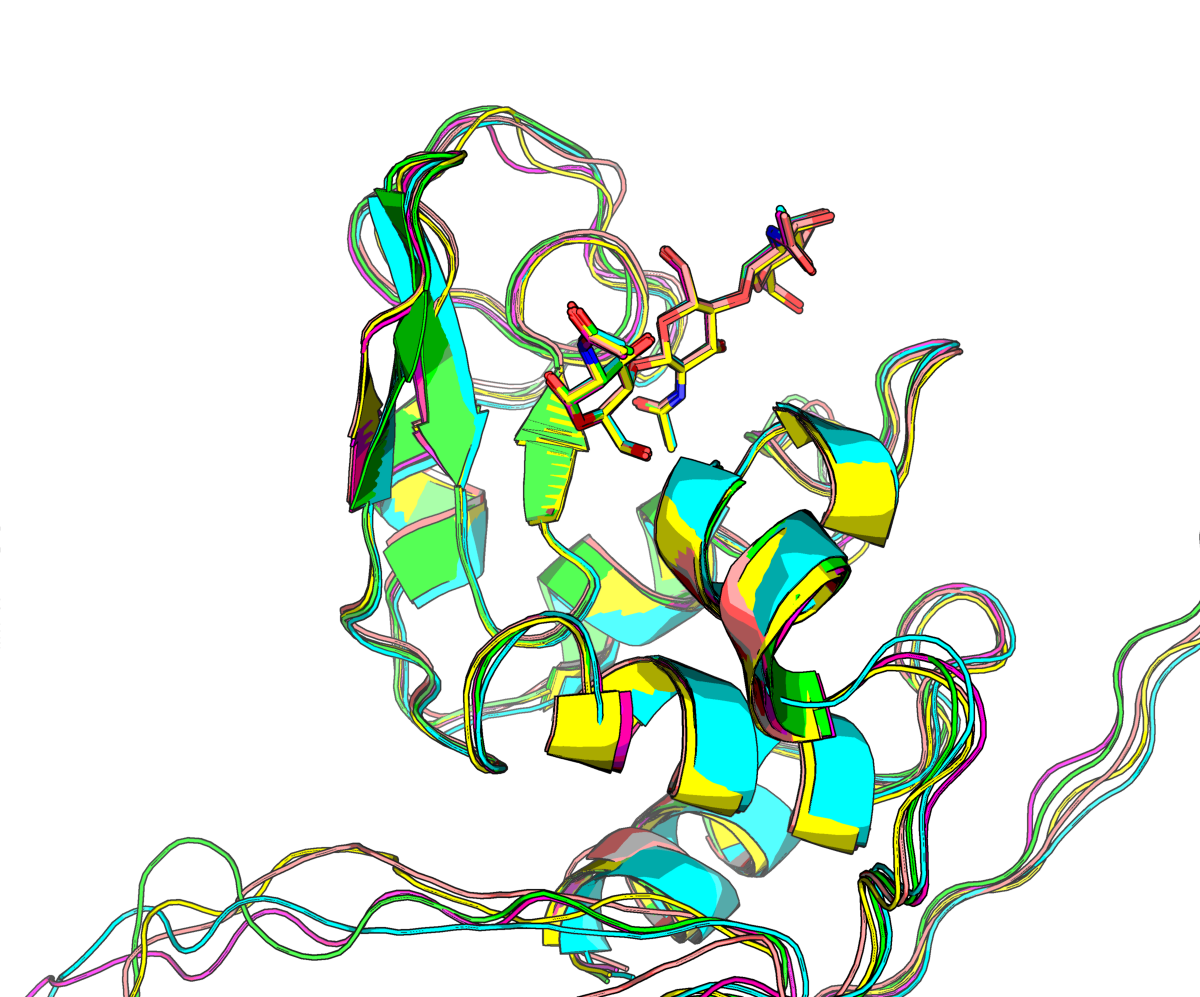 |
| Рис.2. Зона контакта между белком и лигандом в структуре вирусного лизоцима |
Для оценки качества моделей воспользуемся инструментами веб интерфейса WHATIF. По результатам, представленным в таблице (Structure Z-scores, positive is better than average), все модели получились плозими, и выбрать среди них одну самую лучшую сложно. Вероятно, такое низкое качество моделей можно объяснить слишком короткой длиной полипептидной цепи образца.
| Feature/Model | 1 | 2 | 3 | 4 | 5 | Resolution read from PDB file | -1.000 | -1.000 | -1.000 | -1.000 | -1.000 | 1st generation packing quality | -4.996 | -4.825 | -4.906 | -5.160 | -5.192 | 2nd generation packing quality | -6.067 | -6.223 | -5.907 | -6.135 | -6.241 | Ramachandran plot appearance | -1.449 | -1.389 | -1.661 | -1.475 | -1.672 | chi-1/chi-2 rotamer normality | -1.688 | -2.492 | -1.973 | -2.306 | -1.284 | Backbone conformation | -1.562 | -1.530 | -1.384 | -1.629 | -1.856 | Inside/Outside distribution | 1.176 | 1.181 | 1.161 | 1.178 | 1.155 |
|---|
Скрипт для получени картинок в PyMol - script.pml.