Докинг низкомолекулярных лигандов в структуру белка
Для работы был взят лизоцим бактериофага PS119 -
LYS_BPPS1.
Программе Autodock Vina для докинга необходимы специально форматированные файлы pdb
c зарядами и указанием торсионных углов.
Докинг NAG
Подготовка файла NAG
Из банка PDB скачаем SMILES нотацию для NAG -> nag.smi
CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O nag
obgen nag.smi > nag.mol
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Откроем полученную структуру в PyMol и сохраним как .pdb.
С помощью openbabel переформатируем координаты в mol формате во входной файл для Mopac:
babel -ipdb nag.pdb -omop nag_opt_pm6.mop -xk "PM6"Запустим Mopac для оптимизации:
MOPAC2009.exe nag_opt_pm6.mop babel -imopout nag_opt_pm6.out -opdb nag_opt_pm6.pdb
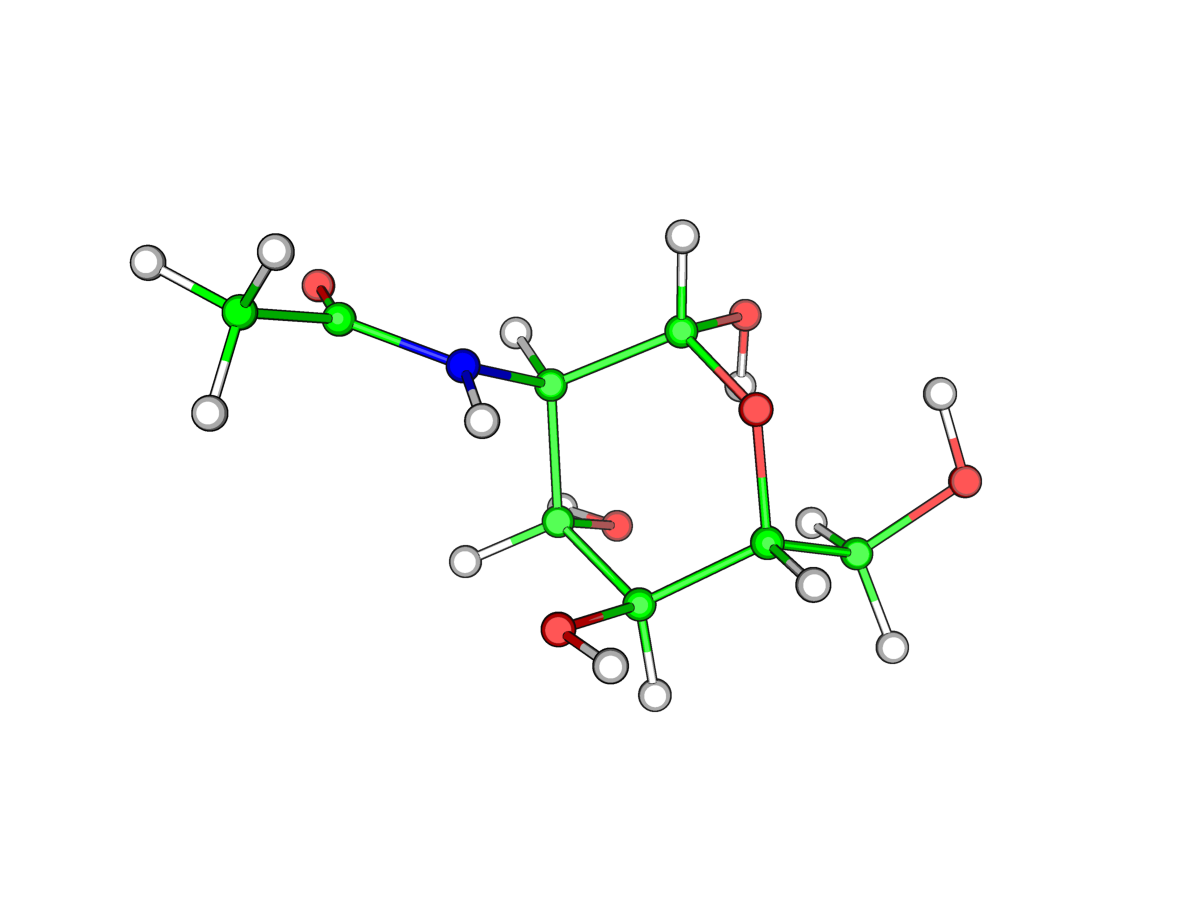 |
| Рис.1. Структура NAG (N-ACETYL-D-GLUCOSAMINE) из файла nag_opt_pm6.pdb |
Скриптом prepare_ligand4.py из пакета Autodock tools создадим pdbqt файл вашего лиганда.
export PATH=${PATH}:/home/preps/golovin/progs/bin
prepare_ligand4.py -l nag_opt_pm6.pdb -o nag.pdbqt
Подготовка файла с белком
Так же, скриптом prepare_receptor4.py из пакета Autodock tools создадим pdbqt файл белка LYS_BPPS1.
prepare_receptor4.py -r model1.pdb -o model1.pdbqt
Параметры докинга
Создадим файл с параметрами докинга vina.cfg.
Область структуры белка, в которой будет происходить поиск места для связывания,
удобно задать как куб с неким центром. Координаты центра мы определим из модели комплекса,
с прошлого занятия (PyMol, команда pseudoatom).
Получаем координаты, которые записываем в файл vina.cfg
[36.562774658203125, 42.758872985839844, 21.227760314941406]
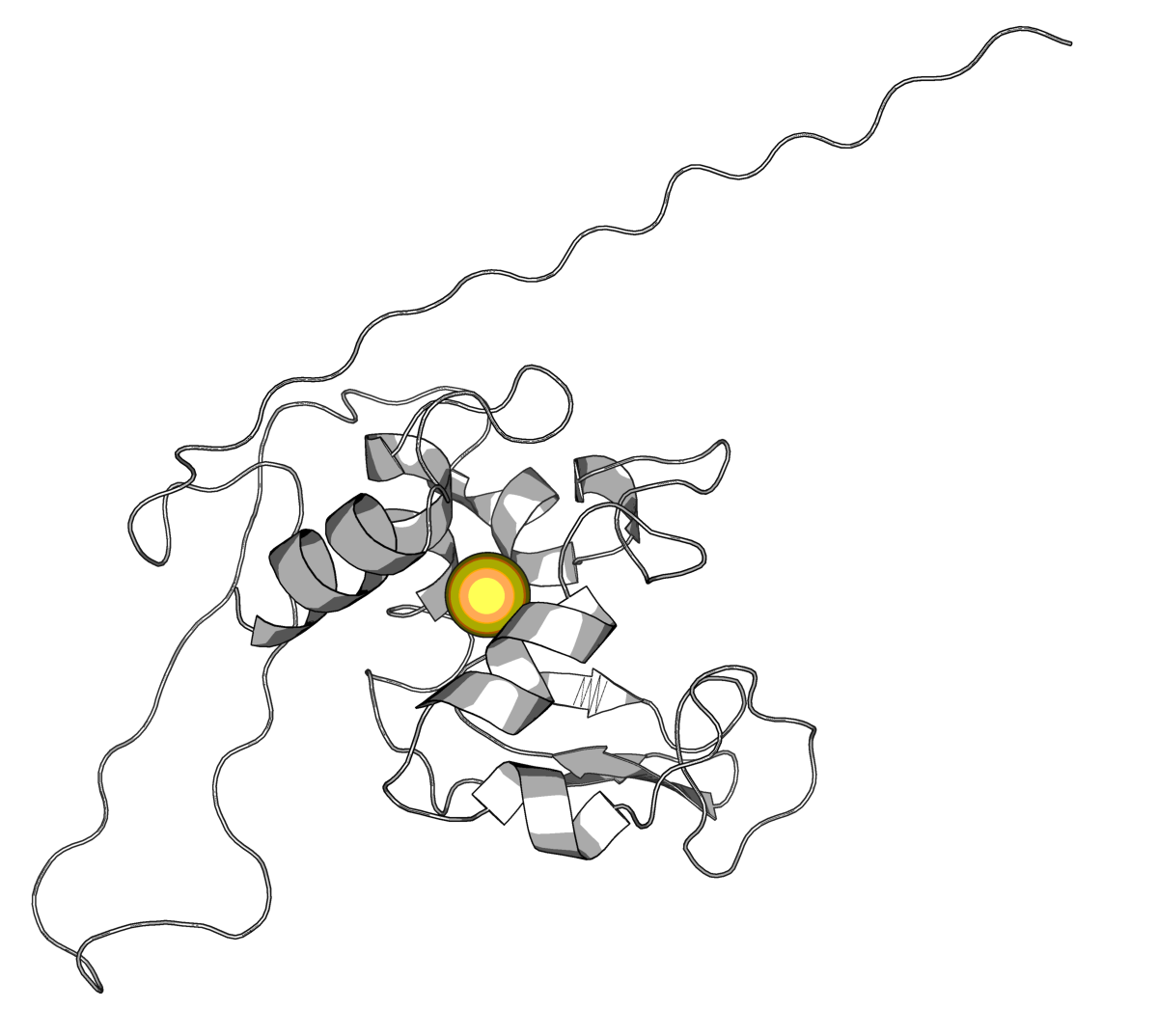 |
| Рис.2. Визуализация модели_1 из прошлого занятия с обозначенным центром масс |
Проведение докинга
vina --config vina.cfg --receptor model1.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.logИз файла nag_prot.log выберем энергии трех лучших расположений и геометрическую разницу между ними.
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.4 0.000 0.000
2 -5.2 11.221 12.589
3 -5.1 2.569 4.989
В PyMol загрузим файлы nag_prot.pdbqt и prot.pdbqt и отобразии все состояния
на одной картинке (Рис.3). Видно, что у лиганда есть свой карман, по которому он гуляет,
но в трех состояних он "убежал".
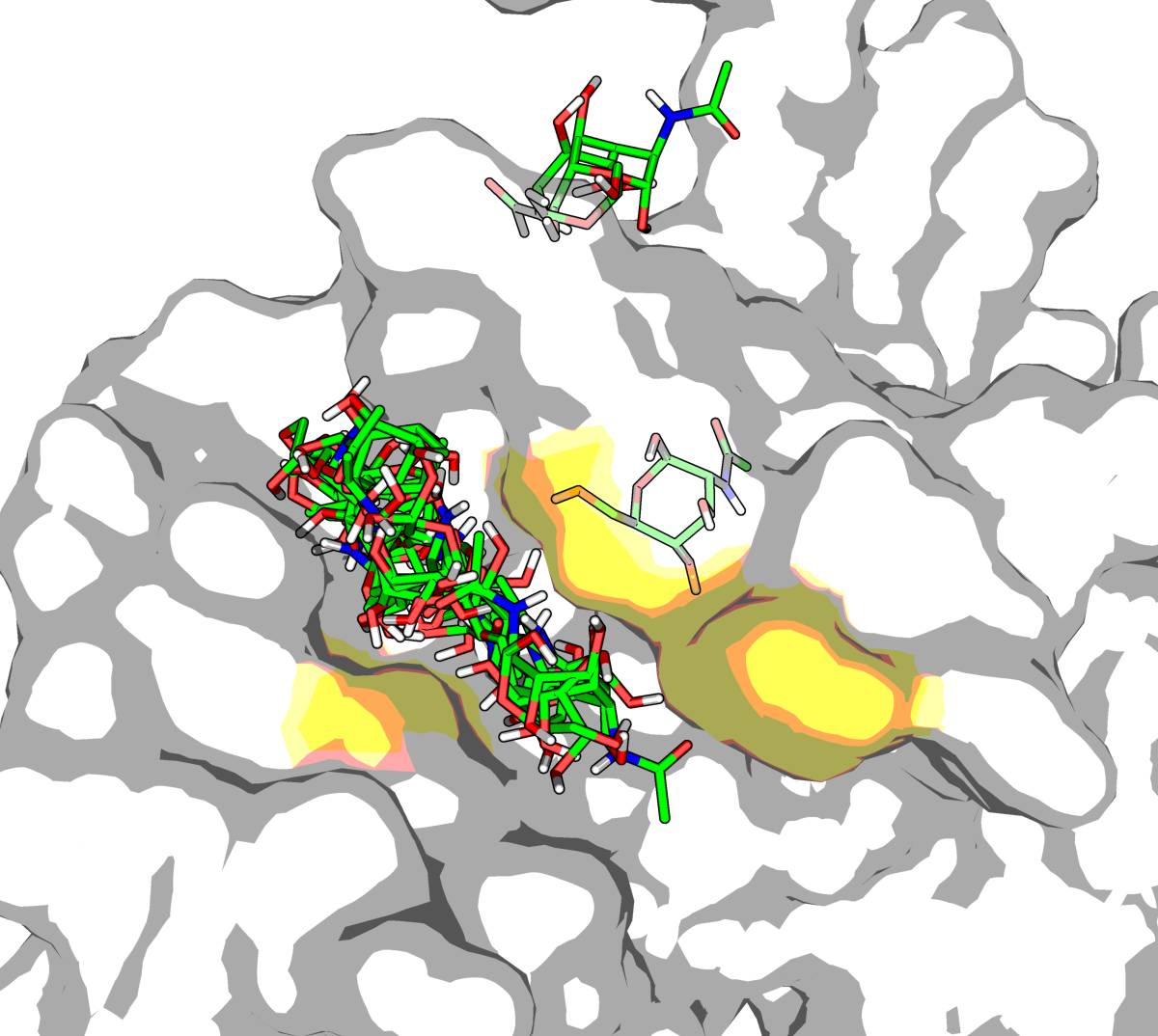 |
| Рис.3. Изображение всех 20 состояний лиганда после докинга |
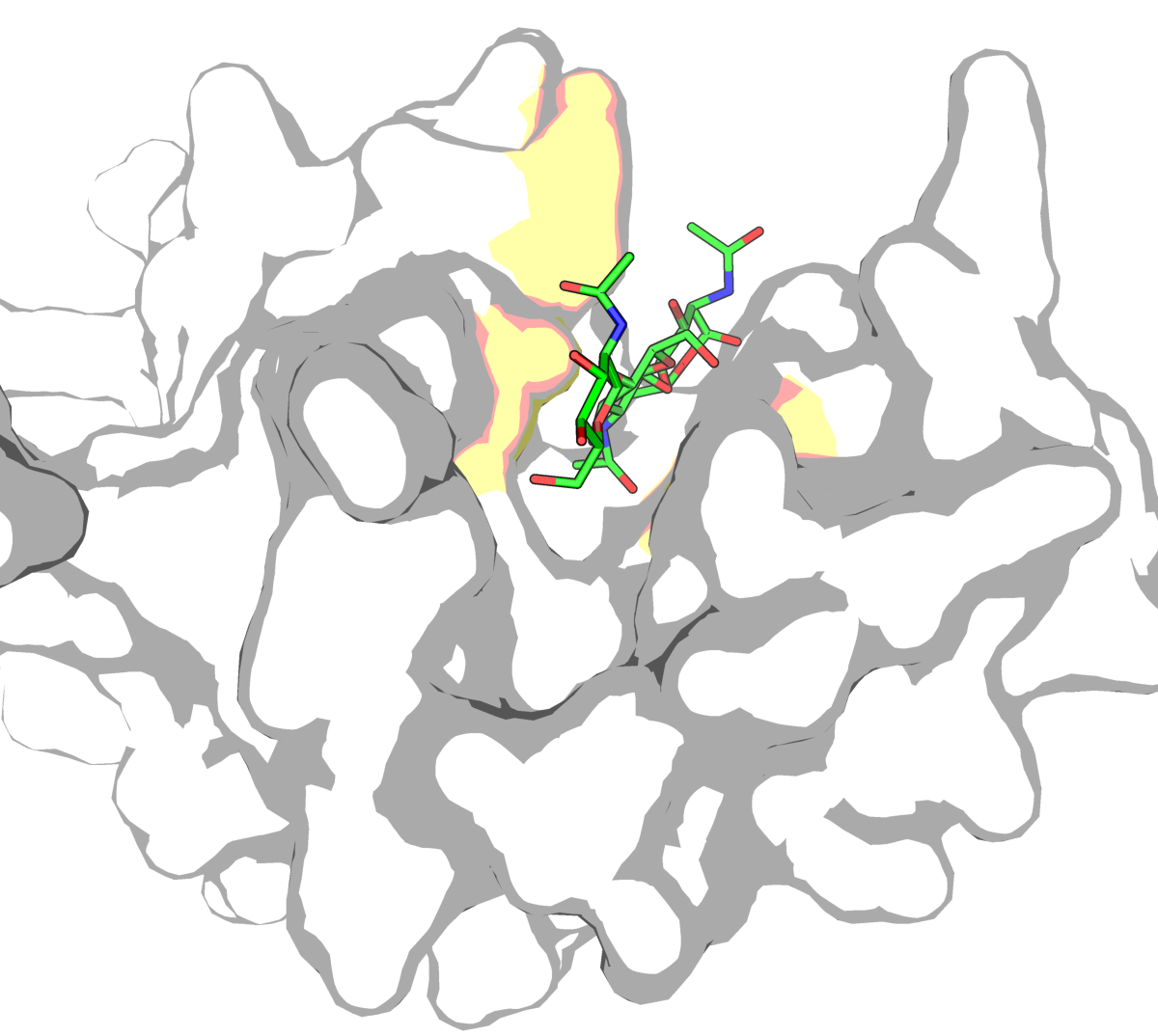
Докинг с учетом подвижности некоторых боковых радикалов белка
Теперь проведём докинг, рассматривая подвижность некоторых боковых радикалов белка. Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты которые использовались в прошлом задании для позиционирования лиганда: CYS54, ALA103 и TYR105 (желтая поверхность на рисунке).
python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py -r model1.pdbqt -s CYS54_ALA103_TYR105 vina --config vina.cfg --receptor model1_rigid.pdbqt --flex model1_flex.pdbqt --ligand nag.pdbqt --out nag_prot_flex.pdbqt --log nag_prot_flex.logИз файла nag_prot_flex.log выберем энергии трех лучших расположений и геометрическую разницу между ними.
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.4 0.000 0.000
2 -5.1 2.064 4.140
3 -5.0 1.213 2.026
Энергия лучшего связывания лиганда с подвижным белком не отличается от такового
с полностью неподвижным (-5.4 ккал/моль).
В случае с подвижным белком лиганд входит в предоставленный ему
"карман" в меньшем количестве состояний.
Докинг с подвижными остатками занял немного больше времени, чем c полностью неподвижным белком.
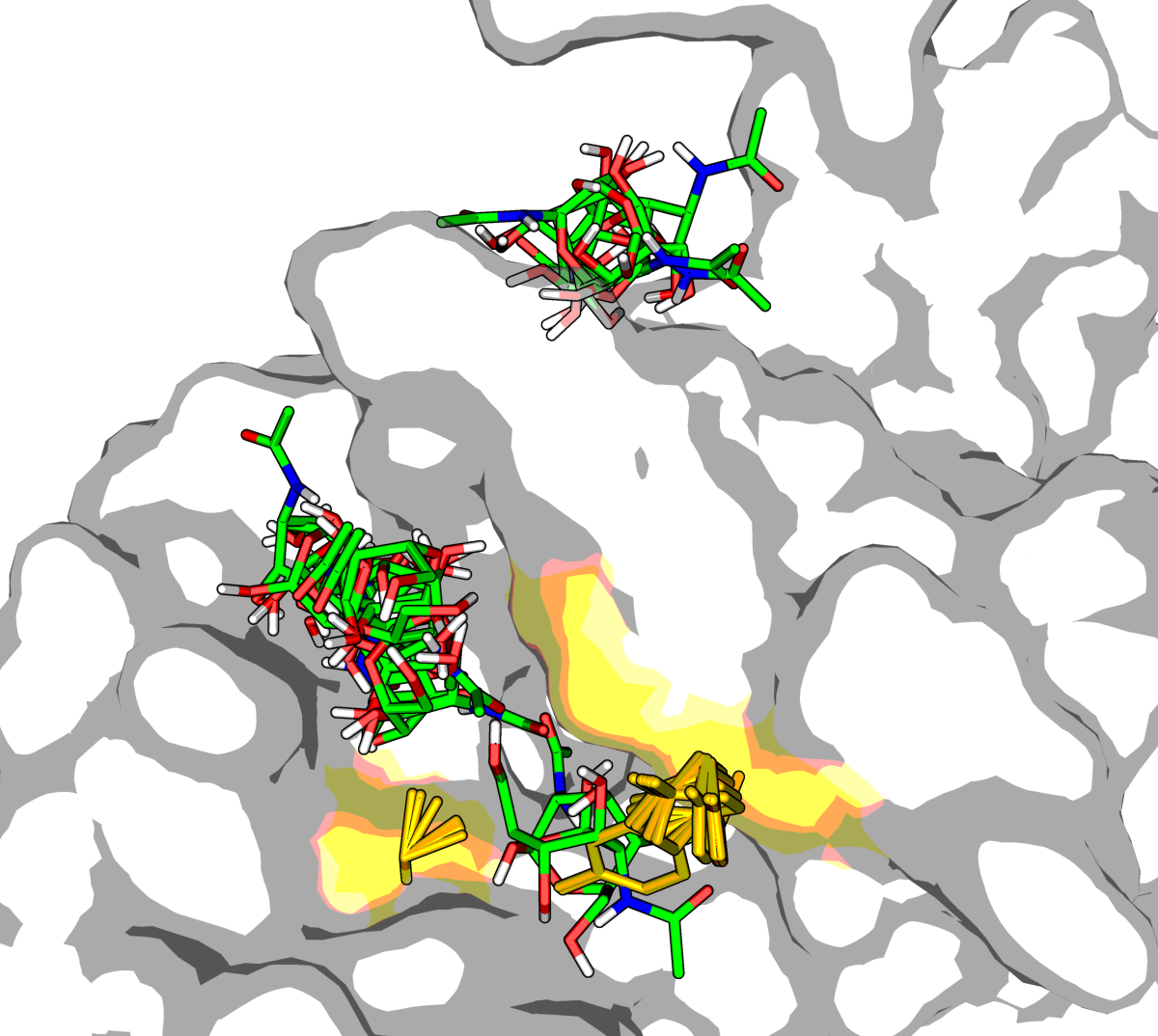 |
| Рис.4. Докинг с учетом подвижности некоторых боковых радикалов белка |
 |
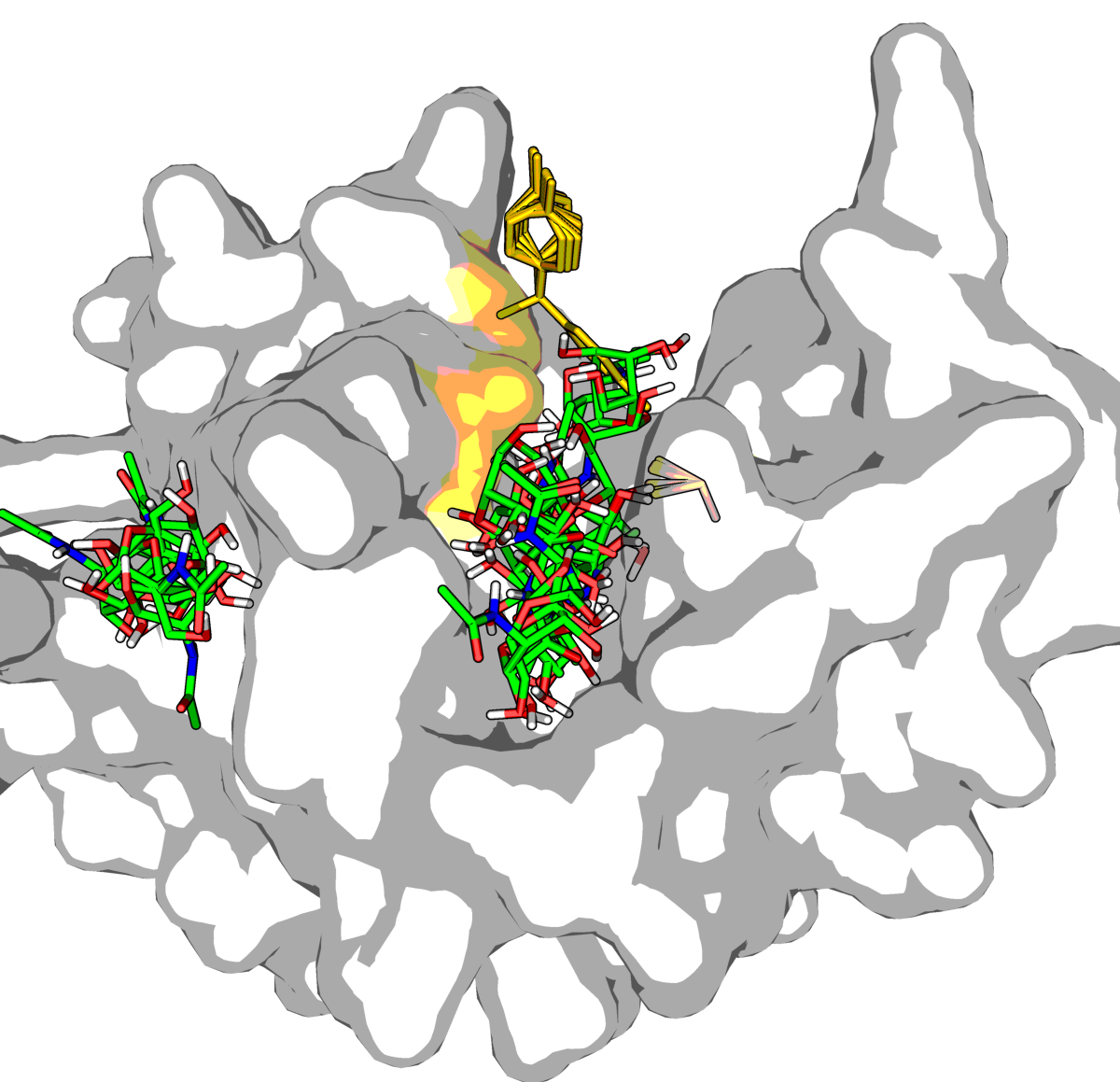 |
| Рис.5. Сравнение глубины попадание лиганда в карман. Слева построенная в предыдущем ДЗ модель, справа - докинг с подвижными боковыми цепями белка. | |
Из Рис.5 можно заключить, что докинг может расположить лиганд наиболее близким образом к старой модели.
Докинг NAG c заменой метильного радикала
Аналогичные операции (обыкновенный докинг) были проделаны еще для четырех лигандов, где СH3C(=O)NH группа была заменена на OH, NH2, H, Ph:
nag2 (OH)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.5 0.000 0.000
2 -4.5 1.444 3.514
3 -4.4 1.271 3.287
nag3 (NH2)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.5 0.000 0.000
2 -5.5 2.046 4.573
3 -5.1 12.436 13.180
nag4 (H)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.7 0.000 0.000
2 -5.4 1.362 3.014
3 -5.0 1.850 3.881
nag5 (Ph)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.3 0.000 0.000
2 -6.3 5.493 9.041
3 -6.1 2.171 5.908
Если судить по энергиям наилучших состояний, лучше всего
с белком связывается Ph-лиганд, хуже всего - OH-лиганд.
Докинг NAG c заменой метильного радикала
Также был проведен докинг с подвижными радикалами белка для четырех лигандов, где СH3C(=O)NH группа была заменена на OH, NH2, Ph:
nag2 (OH)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.5 0.000 0.000
2 -4.4 1.442 3.515
3 -4.3 1.475 2.924
nag3 (NH2)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.1 0.000 0.000
2 -5.1 11.632 13.795
3 -5.1 12.445 14.445
nag5 (Ph)
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.3 0.000 0.000
2 -6.3 5.539 9.082
3 -6.1 2.185 5.905
Энергии лучших состояний существенно не изменились.
Скрипт для получени картинок в PyMol - script.pml.