Главная страница третьего семестра
Предсказание генов во фрагменте генома бактерии Yersinia bercovieri
В этом задании был произведен поиск генов в неанотированном участке
генома бактерии Yersinia bercovieri. Исследовалась часть генома с
идентификатором (ID) AALC01000099 начиная с первого и заканчивая 4000-м
нуклеотидом.
Подготовка
При помощи программы seqret из участка генома Yersinia bercovieri
был вырезан необходимый фрагмент нужной длины.
seqret /home/export/samba/public/tmp/yb.fasta:AALC01000099 sequence.fasta -sask
Извлечение из полученной последовательности открытых рамок
считывания производилось программой getorf. Выбор пал именно
на эту программу, потому что в выходном файле она указывает координаты
рамок в исходной последовательности, а также указывает направление.
getorf piece_gene.fasta -minsize 240 -table bacterial -find 1
Здесь были использованы следующие параметры:
-minsize-указывает минимальную длину рамки считывания
-table-указывет какой генетический код следует использовать
(в нашем случае это стандартный бактериальный код)
-find-объясняет программе, что именно следует считать рамкой
считывания (в нашем случае последовательность, начинающуюся со старт кодона
и заканчивающуюся стоп-кодоном)
В результате выполнения этой программы было найдено 13 рамок
считывания. Эти рамки были импортированны в Exel командой grep
grep AALC01000099 frames_get.fasta > grep2.xls
Далее были созданы индексные файлы BLASTа для всех последовательностей
таксона Enterobacteriales, содержащиеся в банке SwissProt. Было сделано
это при помощи seqret и formatdb.
seqret sw-org:Enterobacteriales
Выходной фаил был назван seqvenses.fasta.
formatdb -i seqvenses.fasta -p T -n seq
Параметры:
-i-входная последовательность
-p-указывает тип последовательности, с которой будет
работать программа (в нашем случае нуклеотидная последовательность)
-n-базовое имя индексных файлов
На этом подготовка была закончена...
Выполнение задания
О том, как создавался скрипт
Смысл всего задания заключался в том, чтобы прогнать полученные рамки
считывания по всем последовательностям из SwissProt (т.е. по аннотированным
последовательностям) таксона Enterobacteriales и посмотреть для каких
из них найдутся гомологи, следовательно такие последовательности мы будем
вправе считать генами. За критерий гомологичности был взят параметр
выравнивания E-value<0,01.
Предположим мы берем одну какую-то рамку считывания...
seqret frames_get.fasta:AALC01000099_1
...и начинаем её BLASTить.
blastall -p blastp -d seq -e 0.01
Чтобы связать рамку считывания с BLASTом, используем
стандартный поток stdout.
seqret frames_get.fasta:AALC01000099_1 stdout | blastall -p blastp -d seq -e 0.01
Полученные данные можно прямиком отправлять в фаил подсчёта результатов,
при помощи всё той же программы grep. Итого, имеем:
seqret frames_get.fasta:AALC01000099_1 stdout | blastall -p blastp -d seq -e 0.01 | grep '^>' -c >>count.txt
Параметр '^>' приводится, чтобы программа читала все строки, начинающиеся с символа >, значек >>
используется, чтобы результат дописывался в конец уже существующего файла.
Такие строчки, при помощи Exel, были созданы для всех рамок считывания. Потом
содержимое документа Exel было скопировано в FAR, отредактировано в нем и
озаглавлено как script.txt.
Вид готового скрипта.
О том, как скрипт работал
При помощи пары несложных команд, скрипт был запущен:
chmod +x script.txt
./script.txt
В результате выполнения скрипта, был получен фаил, в котором
указывалось сколько гомологов к каждой рамке считывания нашел BLAST.
Данные этого файла были приплюсованы к раннее созданной таблице Exel.
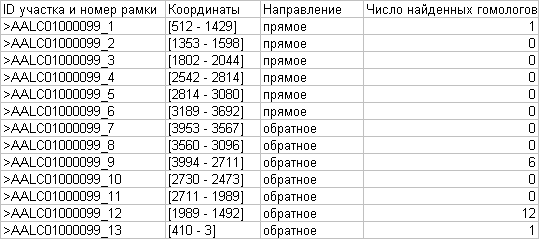
Таким образом, 1, 9, 12 и 13 рамки считывания можно считать генами.
Их схематичное положение изображено ниже:
3<-------410
512------->1429
1492<-------1989
2711<-------3994
©
Низамутдинов Игорь, 2004