На главную страницу третьего семестра
Анализ и визуализация деревьев при помощи програмного пакета PHYLIP
Программы пакета PHYLIP используются для построения и визуализации филогенетических деревьев.
Исследованы два метода восстановления филогенетических деревьев — Bootstrap и Jackknife. Оба они
основаны на том, что из имеющегося выравнивания последовательностей они случайным образом
создают некоторое количество новых и строят не одно, а группу деревьев. После сравнения топологий
этих деревьев методом Maximum Likelihood строится консенсусное дерево, на котором представлены ветви, чаще всего
встречающиеся среди построенных деревьев.
В качестве исходных последовательностей были выбраны листья дерева,
корнем которого является кодирующая нуклеотидная последовательность белка PHOQ_ECOLI.
Bootstrap-анализ выравнивания мутированных последовательностей, соответствующих листьям дерева
Алгоритм Bootstrap основан на том, что новые выравнивания создаются из исходного путём замены 50% позиций в исходном выравнивании
на другие столбцы этого выравнивания (программа seqboot.exe). По полученным выравниваниям было построено 100 деревьев, причём значение
отношения транзиций и трансверсий было выбрано равным 1 (программа dnaml.exe). Затем было создано консенсусное дерево
(программа consence.exe), которое представлено ниже.
+--------------------Mut C
|
+100.0-| +------Mut F
| | +-60.0-|
| +100.0-| +------Mut A
+------| |
| | +-------------Mut B
| |
| +---------------------------Mut E
|
+----------------------------------Mut D
|
Примечание: на внутренних ветвях консенсусного дерева указан процент встречаемости их в 100 построенных деревьях (бутстреп-значения).
Jackknife-анализ выравнивания мутированных последовательностей, соответствующих листьям дерева
Методом Jackknife из исходного выравнивания случайным образом вырезает половину ветвей — так появляются новые
выравнивания, на основании которых строится консенсусное дерево. Использованы те же программы, что и для Bootstrap, было построено
100 деревьев, отношение транзиций к трансверсиям выбрано равным единице.
+------Mut E
+100.0-|
+100.0-| +------Mut D
| |
+-59.0-| +-------------Mut C
| |
+------| +--------------------Mut B
| |
| +---------------------------Mut A
|
+----------------------------------Mut F
|
Ни один из методов не реконструировал дерево с правильной топологией,
однако следует обратить внимание на два факта. Во-первых, в исходном дереве длина ветви
((A,B),(C,D,E,F)) равна всего 7 мут/100нук, что очень мало, поэтому лист F все время
стремится разрушить узел AB. Во-вторых, бутстреп-значения неправильно восстановленных ветвей
тоже малы (60 и 59), что может говорить о недостоверности этого восстановления.
Оба метода не восстановили ветвь ((A,B),все остальное),
бут-стреп значения этой ветви в первом случае, оказалось равным 17, во втором 26.
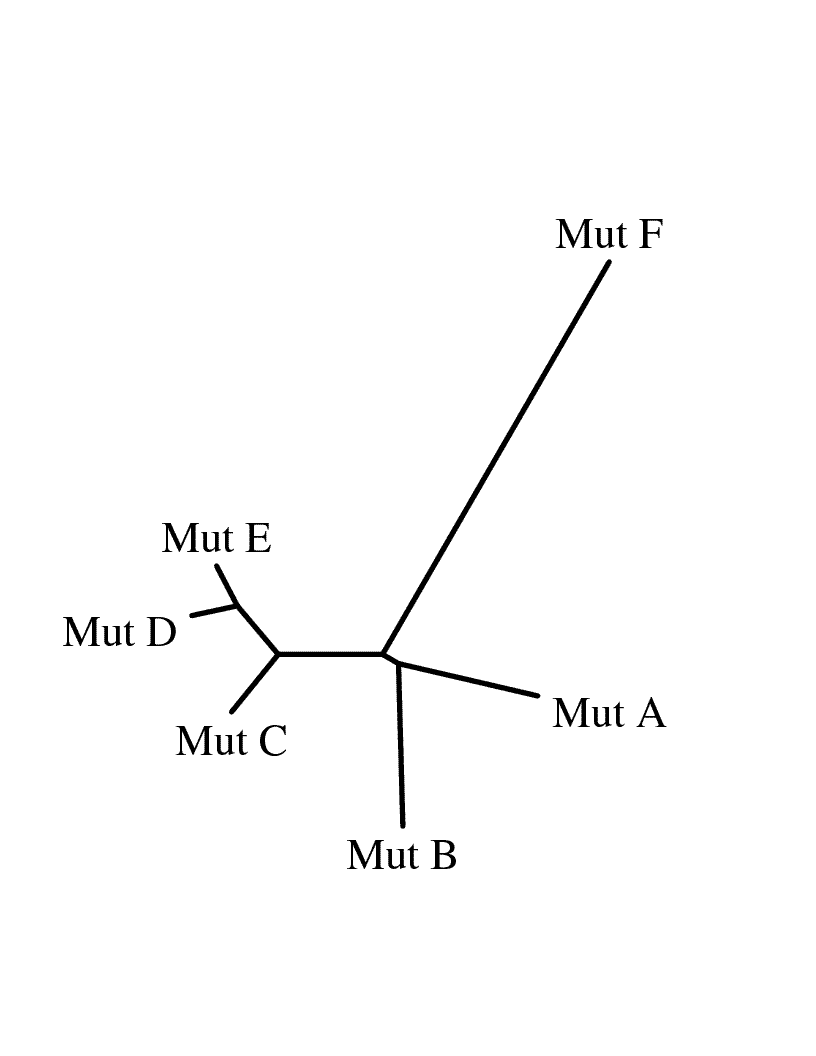
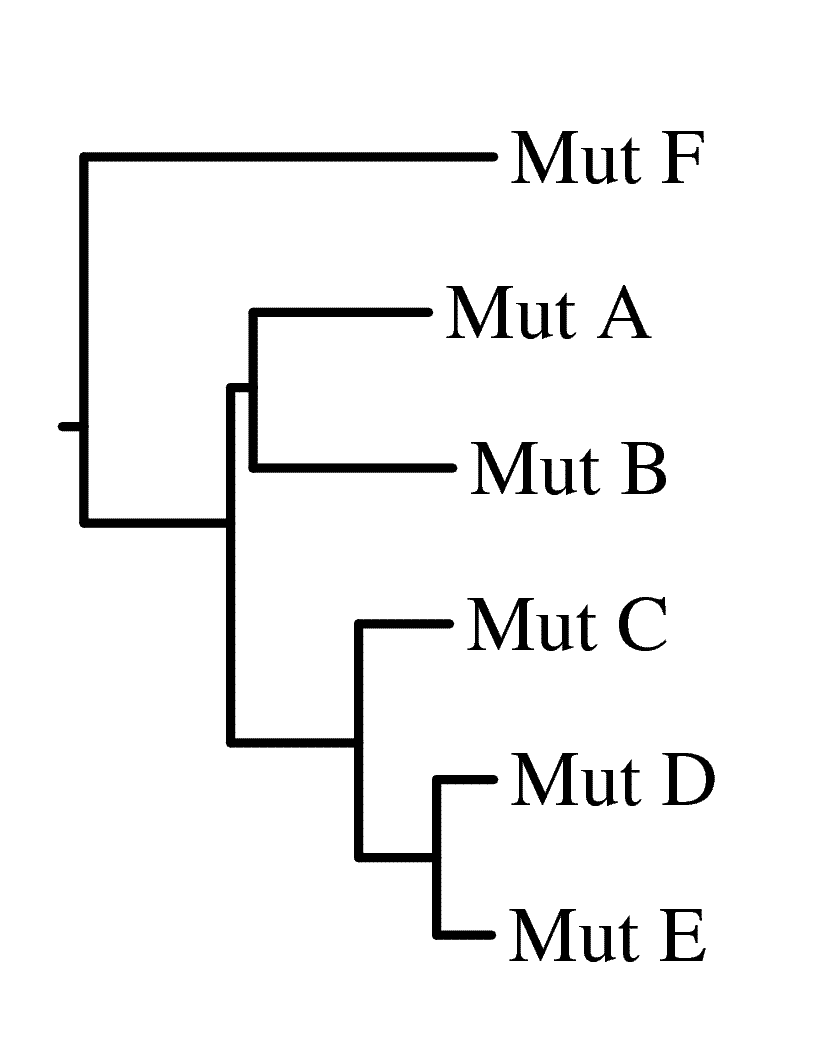
Графические изображения дерева
С помощью программы drawtree.exe создано два графических изображения дерева, полученного методом
Neighbor-Joining: в бескорневом виде и в виде филограммы, ориентированной горизонтально и укоренённой в среднюю точку. Для переукоренения дерева была
использована программа retree.exe.
Ниже представлены полученные изображения деревьев (слева — в бескорневом виде, справа — в виде филограммы).
Восстановление предковой последовательности для выравнивания
Пакет PHYLIP также позволяет восстанавливать предковые последовательности (узлы дерева)
по выравниванию имеющихся (листьев дерева). Была использована программа dnaml.exe.
Получившееся дерево
+-------------------------------------Mut_F
|
| +-----------Mut_B
4--1
| | +------Mut_C
| +---------3
| | +--Mut_E
| +----2
| +---Mut_D
|
+--------Mut_A
|
Выравнивания для узла1, узла 2, узла 3, узла 4 с корнем.
Из представленных выравниваний можно заключить, что наиболее близким к предковой последовательности оказался узел 4
(у него больше процент Identity и Similarity). Оба метода не восстановили ветвь ((A,B),все остальное),
бут-стреп значения этой ветви в первом случае, оказалось равным 17, во втором 26.
Важно заметить, что в некоторых позициях алгоритм не восстанавливает нуклеотид точно. Вместо конкретных нуклеотидов в этих позициях
указывается символ, обозначающий любой из нескольких возможных нуклеотидов. Эти обозначения включены и в матрицу замен, которую использует needle, и из-за наличия
в выравнивании букв, отличных от A, G, C, T, значения для процентов Identity и Similarity отличаются.
Со списком обозначений можно ознакомиться на сайте EBI.
©
Низамутдинов Игорь,2005