Реконструкция деревьев по нуклеотидным последовательностям. Анализ деревьев, содержащих паралоги.
1. Реконструкция деревьев по нуклеотидным последовательностям.
В Uniprot найдем какие-нибудь белки данных бактерий. Посмотрим на какие записи EMBL с полными
геномами они ссылаются. В записях EMBL найдем соответствующие"особенности" (FT), с ключом
(FTkey) "rRNA" и описанием примерно /note="16S rRNA". Полученные данные приведены в таблице:
| Название |
Мнемоника |
AC записи EMBL |
Координаты РНК |
Цепь |
| Bacillus anthracis |
BACAN |
AE016879 |
29129..30635 |
Прямая |
| Bacillus subtilis |
BACsu |
AL009126 |
30279..31832 |
Прямая |
| Clostridium botulinum |
CLOB1 |
CP000726 |
9282..10783 |
Прямая |
| Clostridium tetani |
CLOTE |
AE015927 |
176113..177621 |
Прямая |
| Finegoldia magna |
FINM2 |
AP008971 |
197837..199361 |
Прямая |
| Enterococcus faecalis |
ENTFA |
AE016830 |
1018187..1019708 |
Прямая |
| Geobacillus kaustophilus |
GEOKA |
BA000043 |
30790..32343 |
Прямая |
| Lactobacillus delbrueckii |
LACDA |
CR954253 |
45160..46720 |
Прямая |
| Lactococcus acidophilus |
LACAC |
CP000033 |
59255..60826 |
Прямая |
Далее вырежем нужные участки из записи EMBL командой seqret:
seqret embl:xxxxx -sask
Поместим последовательности в один fasta-файл all.fasta
и отредактируем названия последовательностей, оставив только мнемонику видов.
Создадим выравнивание отобранных белков. Для этого на kodomo запустим программу muscle
с параметрами по умолчанию (т.к. нам не сказано, кк выравнивать) и затем откроем это
выравнивание в JalView.
muscle -in all.fasta -out align.fasta
Выравнивание в fasta-формате.
Далее, построим дерево с помощью программы MEGA (указав Analyze при импорте) методом
Neighbor joining (меню "Phylogeny"):
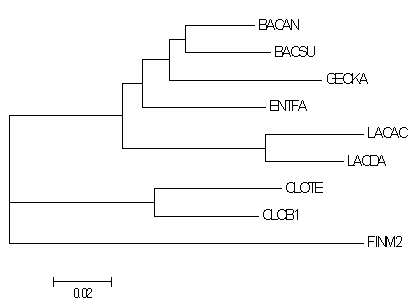 Правильное дерево
Правильное дерево
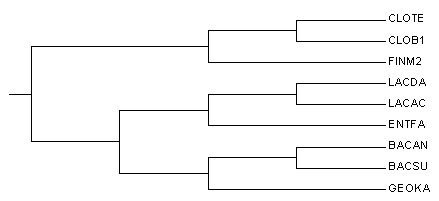 Это дерево содержит 1 нетривиальных ветвей, которых нет в правильное дерево:
Это дерево содержит 1 нетривиальных ветвей, которых нет в правильное дерево:
ветвь {BACAN,BACSU,GEOKA,ENTFA} против {CLOTE,CLOB1,LACDA,LACAC,FINM2},
Это дерево не содержит 1 нетривиальных ветвей, что и правильное дерево:
ветвь {LACDA,LACAC,ENTFA} против {CLOTE,CLOB1,BACAN,BACSU,GEOKA,FINM2},
Видно, что дерево похоже на правильное, но с ним не совподает. Я ожидала, что качество построения по rRNA будет
хуже, чем по белкам. Потому что кодировать одну аминокислоту можно разными триплетами, и
потом вероятность "ошибки" в нуклеотидных последовательностях больше.
1. Построение и анализ дерева, содержащего паралоги
Нашла в своих бактериях достоверные гомологи белка CLPX_BACSU.
Чтобы найти гомологов в заданных организмах, воспользовалась файлом proteo.fasta на диске P, там лежат записи банка UNIPROT, относящиеся к бактериям, перечисленным в таблице к заданию 1.
Провела поиск программой blastp гомологов (с разумным порогом на E-value, скажем, 0,001) и отобрала по мнемонике видов только те находки, которые относятся к отобранным мною бактериям.
Сначала проиндексировала файл:
makeblastdb -in proteo.fasta -out prot -dbtype prot
Затем использовала blastp, выравнивая белки с заготовленной заранее fasta белка CLPX_BACSU
blastp -query clpx_bacsu.fasta -db prot -out prot_clpx.txt -evalue 0.001
Получила набор белков prot_clpx.txt
И создала файл, содержащий только те находки, которые относятся к отобранным мною бактериям.
clpx_homologs.fasta
Полученный файл с выравниванием импортировала в программу MEGA(использовала Neighbor-Joining).
Полученное дерево
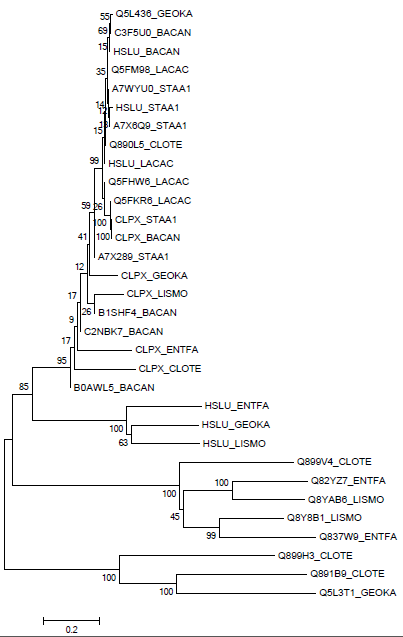 Два гомологичных белка будем называть ортологами, если они а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования.
Два гомологичных белка из одного организма будем называть паралогами.
Ортологи: HSLU_GEOKA и HSLU_LISMO, CLPX_STAA1 и CLPX_BACAN.
Некоторые паралоги:Q82YZ7_ETFA и Q837W9_ENTFA, Q899H3_CLOTE и Q891B9_CLOTE, Q5FHW6_LACAC и Q5FKR6_LACAC.
Два гомологичных белка будем называть ортологами, если они а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования.
Два гомологичных белка из одного организма будем называть паралогами.
Ортологи: HSLU_GEOKA и HSLU_LISMO, CLPX_STAA1 и CLPX_BACAN.
Некоторые паралоги:Q82YZ7_ETFA и Q837W9_ENTFA, Q899H3_CLOTE и Q891B9_CLOTE, Q5FHW6_LACAC и Q5FKR6_LACAC.
© Julia Chudakova