Please note that you are looking at an abridged version of the output (all checks that gave normal results have been removed from this report). You can have a look at the Full report instead.
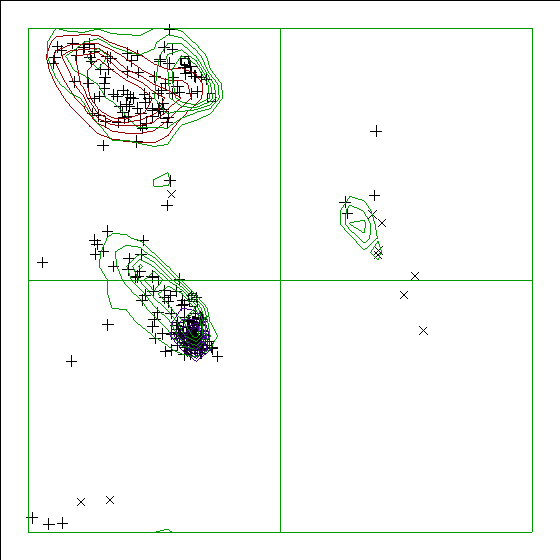
In a colour picture, the residues that are part of a helix are shown in blue, strand residues in red. Preferred regions for helical residues are drawn in blue, for strand residues in red, and for all other residues in green. A full explanation of the Ramachandran plot together with a series of examples can be found at the WHAT_CHECK website.
Obviously, the temperature at which the X-ray data was collected has some importance too:
Crystal temperature (K) :100.000
248 PHE ( 248-) A
RMS Z-score for bond lengths: 0.259
RMS-deviation in bond distances: 0.012
67 ALA ( 67-) A N CA C 126.59 5.5 68 LEU ( 68-) A N CA C 85.50 -9.2 172 VAL ( 172-) A N CA C 99.33 -4.2
RMS Z-score for bond angles: 0.650
RMS-deviation in bond angles: 1.454
68 LEU ( 68-) A 10.42 67 ALA ( 67-) A 7.29 180 ALA ( 180-) A 5.73 69 VAL ( 69-) A 4.52 172 VAL ( 172-) A 4.28 7 GLU ( 7-) A 4.13
These scores give an impression of how `normal' the torsion angles in protein residues are. All torsion angles except omega are used for calculating a `normality' score. Average values and standard deviations were obtained from the residues in the WHAT IF database. These are used to calculate Z-scores. A residue with a Z-score of below -2.0 is poor, and a score of less than -3.0 is worrying. For such residues more than one torsion angle is in a highly unlikely position.
70 PHE ( 70-) A -2.8 108 THR ( 108-) A -2.3 242 VAL ( 242-) A -2.2 204 THR ( 204-) A -2.2 202 ASP ( 202-) A -2.2 185 ILE ( 185-) A -2.1
Residues with `forbidden' phi-psi combinations are listed, as well as residues with unusual omega angles (deviating by more than 3 sigma from the normal value). Please note that it is normal if about 5 percent of the residues is listed here as having unusual phi-psi combinations.
67 ALA ( 67-) A Poor phi/psi 70 PHE ( 70-) A Poor phi/psi 202 ASP ( 202-) A Poor phi/psi 206 ASP ( 206-) A Poor phi/psi 240 SER ( 240-) A Poor phi/psi chi-1/chi-2 correlation Z-score : -0.085
It is not necessarily an error if a few residues have rotamer values below 0.3, but careful inspection of all residues with these low values could be worth it.
121 SER ( 121-) A 0.36
For this check, backbone conformations are compared with database structures using C-alpha superpositions with some restraints on the backbone oxygen positions.
A residue mentioned in the table can be part of a strange loop, or there might be something wrong with it or its directly surrounding residues. There are a few of these in every protein, but in any case it is worth looking at!
10 THR ( 10-) A 0 11 LEU ( 11-) A 0 13 ASN ( 13-) A 0 14 SER ( 14-) A 0 36 MSE ( 36-) A 0 37 ASP ( 37-) A 0 38 LYS ( 38-) A 0 45 ASP ( 45-) A 0 46 HIS ( 46-) A 0 57 THR ( 57-) A 0 58 HIS ( 58-) A 0 64 ALA ( 64-) A 0 66 ASN ( 66-) A 0 67 ALA ( 67-) A 0 68 LEU ( 68-) A 0 69 VAL ( 69-) A 0 70 PHE ( 70-) A 0 71 PHE ( 71-) A 0 72 ASN ( 72-) A 0 89 MSE ( 89-) A 0 90 MSE ( 90-) A 0 109 GLU ( 109-) A 0 135 TYR ( 135-) A 0 152 ASN ( 152-) A 0 164 VAL ( 164-) A 0And so on for a total of 85 lines.
Standard deviation of omega values : 1.671
66 ASN ( 66-) A 1.63
The contact distances of all atom pairs have been checked. Two atoms are said to `bump' if they are closer than the sum of their Van der Waals radii minus 0.40 Angstrom. For hydrogen bonded pairs a tolerance of 0.55 Angstrom is used. The first number in the table tells you how much shorter that specific contact is than the acceptable limit. The second distance is the distance between the centres of the two atoms. Although we believe that two water atoms at 2.4 A distance are too close, we only report water pairs that are closer than this rather short distance.
The last text-item on each line represents the status of the atom pair. If the final column contains the text 'HB', the bump criterion was relaxed because there could be a hydrogen bond. Similarly relaxed criteria are used for 1-3 and 1-4 interactions (listed as 'B2' and 'B3', respectively). BL indicates that the B-factors of the clashing atoms have a low B-factor thereby making this clash even more worrisome. INTRA and INTER indicate whether the clashes are between atoms in the same asymmetric unit, or atoms in symmetry related asymmetric units, respectively.
223 GLY ( 223-) A N <-> 281 HOH ( 568 ) A O 0.39 2.31 INTRA 219 TRP ( 219-) A O <-> 281 HOH ( 568 ) A O 0.25 2.15 INTRA 1 MSE ( 1-) A N <-> 281 HOH ( 489 ) A O 0.23 2.47 INTRA 222 GLN ( 222-) A C <-> 281 HOH ( 568 ) A O 0.18 2.62 INTRA 218 VAL ( 218-) A CG2 <-> 281 HOH ( 568 ) A O 0.18 2.62 INTRA 203 ASP ( 203-) A CB <-> 281 HOH ( 568 ) A O 0.15 2.65 INTRA 146 HIS ( 146-) A ND1 <-> 148 GLN ( 148-) A N 0.13 2.87 INTRA BL 58 HIS ( 58-) A O <-> 61 LYS ( 61-) A NZ 0.11 2.59 INTRA BL 242 VAL ( 242-) A CG2 <-> 243 VAL ( 243-) A N 0.10 2.90 INTRA BL 176 PHE ( 176-) A O <-> 180 ALA ( 180-) A N 0.09 2.61 INTRA BL 170 GLY ( 170-) A N <-> 197 PHE ( 197-) A CE2 0.09 3.01 INTRA BL 222 GLN ( 222-) A N <-> 281 HOH ( 568 ) A O 0.05 2.65 INTRA 241 ASP ( 241-) A OD1 <-> 242 VAL ( 242-) A N 0.05 2.55 INTRA 93 CYS ( 93-) A O <-> 97 GLY ( 97-) A N 0.04 2.66 INTRA BL 39 LEU ( 39-) A N <-> 40 PRO ( 40-) A CD 0.02 2.98 INTRA BL 195 LYS ( 195-) A NZ <-> 281 HOH ( 509 ) A O 0.01 2.69 INTRA 109 GLU ( 109-) A N <-> 110 GLN ( 110-) A N 0.01 2.59 INTRA BL 45 ASP ( 45-) A CG <-> 46 HIS ( 46-) A CE1 0.01 3.19 INTRA 29 ASP ( 29-) A OD1 <-> 59 HIS ( 59-) A CE1 0.01 2.79 INTRA BL 179 HIS ( 179-) A CD2 <-> 281 HOH ( 406 ) A O 0.01 2.79 INTRA BL
The packing environment of the residues is compared with the average packing environment for all residues of the same type in good PDB files. A low packing score can indicate one of several things: Poor packing, misthreading of the sequence through the density, crystal contacts, contacts with a co-factor, or the residue is part of the active site. It is not uncommon to see a few of these, but in any case this requires further inspection of the residue.
222 GLN ( 222-) A -5.96 134 PRO ( 134-) A -5.62 179 HIS ( 179-) A -5.57 111 LYS ( 111-) A -5.04
The table below lists the first and last residue in each stretch found, as well as the average residue Z-score of the series.
208 PRO ( 208-) A - 211 PHE ( 211-) A -1.63
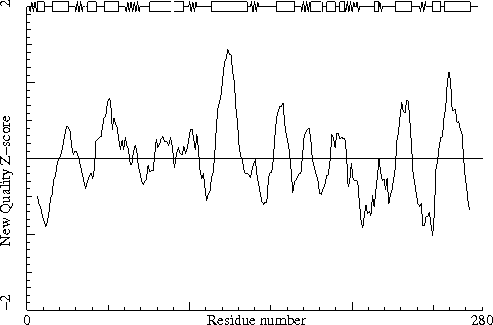
80 ASN ( 80-) A 179 HIS ( 179-) A
Hydrogen bond donors that are buried inside the protein normally use all of their hydrogens to form hydrogen bonds within the protein. If there are any non hydrogen bonded buried hydrogen bond donors in the structure they will be listed here. In very good structures the number of listed atoms will tend to zero.
Waters are not listed by this option.
12 GLU ( 12-) A N 13 ASN ( 13-) A ND2 37 ASP ( 37-) A N 64 ALA ( 64-) A N 66 ASN ( 66-) A ND2 107 VAL ( 107-) A N 143 PHE ( 143-) A N 153 THR ( 153-) A N 181 MSE ( 181-) A N 202 ASP ( 202-) A N 248 PHE ( 248-) A N Only metal coordination for 142 GLU ( 142-) A OE2 Only metal coordination for 174 ASP ( 174-) A OD2 Only metal coordination for 200 HIS ( 200-) A ND1
Side-chain hydrogen bond acceptors buried inside the protein normally form hydrogen bonds within the protein. If there are any not hydrogen bonded in the optimized hydrogen bond network they will be listed here.
Waters are not listed by this option.
7 GLU ( 7-) A OE2 202 ASP ( 202-) A OD2
45 ASP ( 45-) A H-bonding suggests Asn 270 ASP ( 270-) A H-bonding suggests Asn
The second part of the table mostly gives an impression of how well the model conforms to common refinement restraint values. The first part of the table shows a number of global quality indicators.
Structure Z-scores, positive is better than average:
1st generation packing quality : 0.974 2nd generation packing quality : -0.200 Ramachandran plot appearance : -0.064 chi-1/chi-2 rotamer normality : -0.085 Backbone conformation : 0.196
Bond lengths : 0.259 (tight) Bond angles : 0.650 (tight) Omega angle restraints : 0.304 (tight) Side chain planarity : 0.254 (tight) Improper dihedral distribution : 0.549 B-factor distribution : 0.782 Inside/Outside distribution : 0.951
The second part of the table mostly gives an impression of how well the model conforms to common refinement restraint values. The first part of the table shows a number of global quality indicators, which have been calibrated against structures of similar resolution.
Resolution found in PDB file : 1.80
Structure Z-scores, positive is better than average:
1st generation packing quality : 1.3 2nd generation packing quality : -0.6 Ramachandran plot appearance : 0.0 chi-1/chi-2 rotamer normality : 0.2 Backbone conformation : -0.0
Bond lengths : 0.259 (tight) Bond angles : 0.650 (tight) Omega angle restraints : 0.304 (tight) Side chain planarity : 0.254 (tight) Improper dihedral distribution : 0.549 B-factor distribution : 0.782 Inside/Outside distribution : 0.951 ==============
WHAT IF
G.Vriend,
WHAT IF: a molecular modelling and drug design program,
J. Mol. Graph. 8, 52--56 (1990).
WHAT_CHECK (verification routines from WHAT IF)
R.W.W.Hooft, G.Vriend, C.Sander and E.E.Abola,
Errors in protein structures
Nature 381, 272 (1996).
(see also http://swift.cmbi.ru.nl/gv/whatcheck for a course and extra inform
Bond lengths and angles, protein residues
R.Engh and R.Huber,
Accurate bond and angle parameters for X-ray protein structure
refinement,
Acta Crystallogr. A47, 392--400 (1991).
Bond lengths and angles, DNA/RNA
G.Parkinson, J.Voitechovsky, L.Clowney, A.T.Bruenger and H.Berman,
New parameters for the refinement of nucleic acid-containing structures
Acta Crystallogr. D52, 57--64 (1996).
DSSP
W.Kabsch and C.Sander,
Dictionary of protein secondary structure: pattern
recognition of hydrogen bond and geometrical features
Biopolymers 22, 2577--2637 (1983).
Hydrogen bond networks
R.W.W.Hooft, C.Sander and G.Vriend,
Positioning hydrogen atoms by optimizing hydrogen bond networks in
protein structures
PROTEINS, 26, 363--376 (1996).
Matthews' Coefficient
B.W.Matthews
Solvent content of Protein Crystals
J. Mol. Biol. 33, 491--497 (1968).
Protein side chain planarity
R.W.W. Hooft, C. Sander and G. Vriend,
Verification of protein structures: side-chain planarity
J. Appl. Cryst. 29, 714--716 (1996).
Puckering parameters
D.Cremer and J.A.Pople,
A general definition of ring puckering coordinates
J. Am. Chem. Soc. 97, 1354--1358 (1975).
Quality Control
G.Vriend and C.Sander,
Quality control of protein models: directional atomic
contact analysis,
J. Appl. Cryst. 26, 47--60 (1993).
Ramachandran plot
G.N.Ramachandran, C.Ramakrishnan and V.Sasisekharan,
Stereochemistry of Polypeptide Chain Conformations
J. Mol. Biol. 7, 95--99 (1963).
Symmetry Checks
R.W.W.Hooft, C.Sander and G.Vriend,
Reconstruction of symmetry related molecules from protein
data bank (PDB) files
J. Appl. Cryst. 27, 1006--1009 (1994).
Ion Checks
I.D.Brown and K.K.Wu,
Empirical Parameters for Calculating Cation-Oxygen Bond Valences
Acta Cryst. B32, 1957--1959 (1975).
M.Nayal and E.Di Cera,
Valence Screening of Water in Protein Crystals Reveals Potential Na+
Binding Sites
J.Mol.Biol. 256 228--234 (1996).
P.Mueller, S.Koepke and G.M.Sheldrick,
Is the bond-valence method able to identify metal atoms in protein
structures?
Acta Cryst. D 59 32--37 (2003).
Checking checks
K.Wilson, C.Sander, R.W.W.Hooft, G.Vriend, et al.
Who checks the checkers
J.Mol.Biol. (1998) 276,417-436.