аннотация
Мотивация: исследуя геном Vibrio Cholerae, можно понять, как свободноживущая бактерия приобрела патогенные свойства. Интересно также, как генетические особенности позволяют совмещать два различных образа жизни: свободноживущий и эндобионтный.
введение
Vibrio cholerae – грамотрицательная, факультативно анаэробная, неинвазивная, патогенная бактерия, вызывающая заболевание желудочно-кишечного тракта – холеру, которая приводит к эпидемиям.
Холера известна с древних времен, первый случай был описан в XVI веке, в XIX веке заболевание распространилось по всему миру, сейчас наиболее часто вспышки возникают в Африке. В истории известны 7 эпидемий холеры [1, 2].
V. cholerae – свободноживущий, плавающий организм (обитает в муссонном климате), широко распространена в прибрежных водах и морепродуктах, при проглатывании которых и происходит заражение. Проходя через желудок, бактерия попадает в тонкий кишечник, на эпителии которого начинают формироваться колонии. Бактерии выделяют в межклеточное пространство холерный токсин – белковый комплекс, который нарушает ионный транспорт эпителиальных клеток, что приводит к потере воды и ионов, а, значит, к диарее, рвоте, мышечным спазмам. В некоторых случаях потеря жидкости может привести к летальному исходу в течение суток [1].
методы
Стандартные данные о геноме раздел 3.1 были получены из файла assembly_stats для сборки с сайта NCBI (1).
Исследование содержания нуклеотидов по репликонам и кодирующим последовательностям раздел 3.2 проводился средствами языка программирования Python в Google Colab (6). В ходе скрипта происходит скачивание и обработка файлов с полными последовательностями для репликонов и файла с записями кодирующих последовательностей с сайта NCBI, после данные из файлов принимает на вход функция average_bases(), которая выдает в качестве результата содержание нуклеотидов. Критерий хи-квадрат был подсчитан с помощью библиотеки scipy.
Типы генов раздел 3.3, а также длины и количества белков раздел 3.7 были подсчитаны в Google Sheets (3, 5) с использованием функции COUNTIFS на основе таблицы локальных особенностей (feature_table.txt) с сайта NCBI (2). Подсчет различных тРНК раздел 3.4 был произведен с помощью фильтров над таблицей локальных особенностей (feature_table) в Google Sheets (3).
Распределение генов по "+" и "-" цепям для репликонов раздел 3.5 было подсчитано средствами Python 3 в Google Colab (6). В ходе скрипта обрабатывается файл с записями кодирующих последовательностей, если в комментарии к записи встречается слово "complement", то скрипт засчитывает эту последовательность, как последовательность на "-" цепи, в обратном случае - как последовательность на "+" цепи. t-тест был проведен с помощью библиотеки scipy.
Старт-кодоны кодирующих последовательностей раздел 3.6 были подсчитаны в Google Sheets (4) с использованием функции VLOOKUP, перед этим они были отобраны средствами командной строки bash.
результаты и обсуждения
3.1 Стандартные данные о геноме V Cholerae
Хромосома 1 содержит большую часть генов, отвечающих за рост и жизнеспособность. На хромосоме 2 также присутствуют метаболистические гены. Более подробное сравнение двух хромосом приводится в последующих результатах.
Плазмида в исследовании практически не затрагивается в связи с нехваткой данных для какого-либо ее анализа.
GC-состав чуть ниже 50%, что свидетельствует о мезофильности бактерии. С одной стороны, генетический материал должен быть легко подвержен изменениям для адаптации к внешней среде при свободном образе жизни, с другой стороны, должен быть достаточно устойчив, чтобы не разрушаться во внутриорганизменной среде.
| Молекула | Длина (п.о.) | GC-состав (%) |
|---|---|---|
| Общее | 4 138 412 | 47.5 |
| Хромосома 1 | 2 948 589 (71%) | 47.5 |
| Хромосома 2 | 1 140 710 (28%) | 47 |
| Плазмида | 49 113 (1%) | 41 |
3.2 Исследование содержания нуклеотидов
Для обеих хромосом выполняются все правила Чаргаффа: количество аденина примерно равно количеству тимина, а количество цитозина – количеству гуанина; количество пуринов равно количеству пиримидинов (A + G = T + C); количество оснований с аминогруппами в положении 6 равно количеству оснований с кетогруппами в положении 6 (A + C = T + G).
Для оценки случайности распределения нуклеотидов можно использовать критерий хи-квадрат, который рассчитывается по формуле:
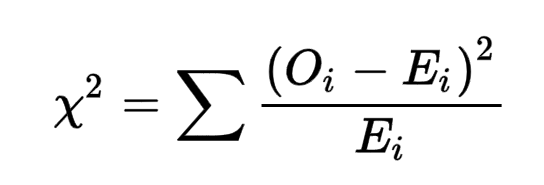
где Oi – наблюдаемое значение, Ei – ожидаемое значение. За ожидаемые значения примем значения нуклеотидного распределения исходя из GC-состава репликонов: для хромосомы 1: %A = %T = 26.25, %C = %G = 23.75; для хромосомы 2: %A = %T = 26.5, %C = %G = 23.5. Тогда значения хи-квадрат равны 0.0017 и 0.0004 для первой и второй хромосом соответственно, а p-value практически равно 1 для обеих хромосом. Таким образом, наблюдаемые данные очень хорошо соответствуют ожидаемым и полностью подтверждают нулевую гипотезу.
| Репликон | A (%) | T (%) | G (%) | C (%) |
|---|---|---|---|---|
| Хромосома 1 | 26.2 | 26.0 | 23.9 | 23.8 |
| Хромосома 2 | 26.5 | 26.6 | 23.4 | 23.5 |
3.3 Типы генов в геноме V Cholerae
На хромосоме 2 достаточно много псевдогенов – генов, похожих на функциональные, но потерявших способность кодировать белок или РНК. Псевдогены возникают в результате мутаций и эволюционных процессов.
Плазмида содержит только белок-кодирующие гены, но большая часть этих белков – гипотетические, т.е. их структура и функции не установлены экспериментально.
Исходя из распределения генов можно предположить, что изначально две хромосомы были одинаковы, а в ходе эволюции важные гены "перетекали" со второй хромосомы на первую в целях более эффективной репликации и реализации генетической информации на одной хромосоме вместо двух. Вторая хромосома не редуцировалась полностью, так как все еще содержит важные функциональные гены.
Другая теория заключается в том, что вторая хромосома обеспечивает эволюционный резерв для жизни в разных экологических нишах.
Некоторые источники свидетельствуют о наличии горизонтального переноса генов, так как на первой и второй хромосоме присутствуют гены одинаковых белков, но филогенетика показывает, что они имеют разное происхождение [3].
| Ген | Хромосома 1 | Хромосома 2 | Плазмида |
|---|---|---|---|
| protein_coding | 2540 | 1025 | 63 |
| tRNA | 98 | 4 | 0 |
| rRNA | 31 | 0 | 0 |
| tmRNA | 1 | 0 | 0 |
| ncRNA | 1 | 0 | 0 |
| RNase_P_RNA | 2 | 0 | 0 |
| SRP_RNA | 2 | 0 | 0 |
| pseudogene | 13 | 26 | 0 |
protein_coding – белок-кодирующие, tRNA – кодирующие транспортные РНК, rRNA – кодирующие рибосомальные РНК, tmRNA – кодирующие транспортно-матричные РНК, ncRNA – кодирующие некодирующие РНК, RNase_P_RNA – кодирующие РНКзу P, SPR_RNA – кодирующие 7SL РНК, pseudogene – псевдогены.
3.4 Распределение числа различных тРНК по их аминокислотам
В таблице 5 отображено распределение тРНК по аминокислотам. Для некоторых аминокислот имеются разные тРНК с одинаковыми антикодонами. Скорее всего, в геноме неоднократно происходили дупликации участков, кодирующих тРНК.
| Аминокислота | Число тРНК | Норма | Аминокислота | Число тРНК | Норма |
|---|---|---|---|---|---|
| Ala | 6 | 2 | Leu | 12 | 3 |
| Arg | 8 | 3 | Lys | 3 | 1 |
| Asn | 4 | 1 | Met | 7 | 2 |
| Asp | 5 | 1 | Phe | 3 | 1 |
| Cys | 3 | 1 | Pro | 4 | 2 |
| Gln | 5 | 1 | Ser | 5 | 3 |
| Glu | 5 | 1 | Thr | 6 | 2 |
| Gly | 9 | 2 | Trp | 1 | 1 |
| His | 2 | 1 | Tyr | 4 | 1 |
| Ile | 5 | 2 | Val | 5 | 2 |
3.5 Распределение генов по цепям
Было подсчитано количество генов на “+” и “-” цепях обеих хромосом, результаты представлены в таблице 6. Их количества приблизительно равны в обоих случаях, что значит, что распределение генов по цепям, скорее всего, случайно.
| Репликон | "+" цепь | "-" цепь |
|---|---|---|
| Хромосома 1 | 1318 | 1235 |
| Хромосома 2 | 567 | 481 |
3.6 Определение старт-кодонов кодирующих последовательностей
В таблице 7 приведено количество встречаемых старт-кодонов на кодирующих цепях.
Типичный старт-кодон ATG встречается в 89% случаев по всем CDS. На нормальных CDS встречаются старт-кодоны, отличающиеся от классического ATG на одно основание, что вызвано точечными мутациями. Наиболее часто это замена A на G (замена с переходом), в два раза реже – замена A на T (замена с трансверсией). На псевдогенах встречается кодоны с заменой и двух, и всех трех азотистых оснований.
| Кодон | Всего | Нормальная кодирующая последовательность | Псевдогены |
|---|---|---|---|
| ATG | 3237 | 16 | 3253 |
| GTG | 252 | 5 | 257 |
| TTG | 123 | 5 | 128 |
| ATC | 7 | 1 | 8 |
| ATT | 4 | 3 | 7 |
| ATA | 3 | 1 | 4 |
| CTG | 2 | 1 | 3 |
| GAT, GCA, CAA, CTC, TAT, TGT, TCA | 0 | х1 | х1 |
| остальное | 0 | 0 | 0 |
3.7 Длины белков, закодированных в геноме V. cholerae
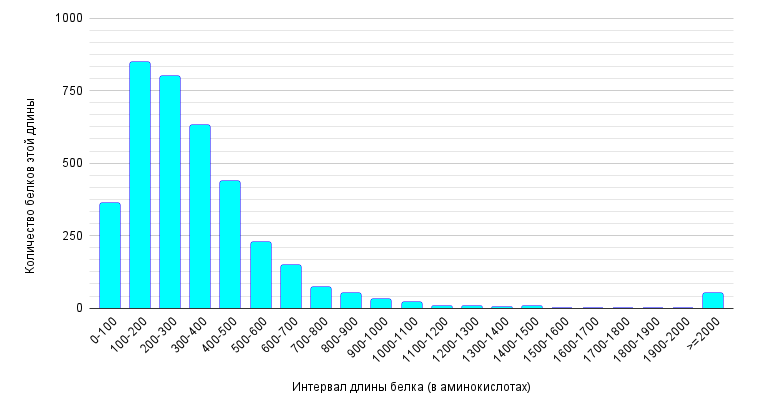
Наибольшее количество белков (442) имеет длину 150-200 аминокислотных остатков. Самый большой белок имеет длину 4545 аминокислотных остатков, этот белок функционально связан с токсином RtxA.
Поскольку гистограмма отображает все возможные белки V. cholerae, её можно назвать отображением популяции. Были вычислены её параметры: среднее значение равно 325, медианное значение равно 277. Среднее значение значительно отличается от медианного, можно сделать вывод, что распределение не является нормальным (подтверждается формой графика, он имеет вид гамма-распределения). Больше половины белков имеют длину менее 300 а.о., также имеются белки очень большой длины (>2000 а.о.), за счет которых среднее значение увеличено относительно медианного.
С помощью гистограммы можно посчитать вероятность встретить белки определенной длины. Таким образом, вероятность встретить белок длиной менее 100 а.о. равна 9.8%, более 1000 а.о. – 1.5%, от 100 до 300 а.о. – 45%.
благодарности
Выражаю сердечную благодарность следующим личностям и сущностям:
сопроводительные материалы и ссылки
(1) NCBI taxonomy browser, txid666
(2) Геномные данные Vibrio cholerae
(3) Таблица генов белков и различных тРНК для репликонов
(4) Таблица с частотой встречаемости старт-кодонов
литература
