



BLAST - это программа, находящаяся на сервере NCBI, которая позволяет находить белковые последовательности в базе данных Swiss-prot, сходные с поданной на вход последовательностью. Используя эту программу, биологи чаще всего хотят найти гомологичные последовательности. BLAST также умеет строить выравнивание двух и более последовательностей относительно друг друга, строить карту локального сходства, дерево выравнивания. BLAST является самой популярной биоинформатической программой, поскольку она проста в использовании и выполняет важные задачи.
Я зарегистрировалась на сайте NCBI, выбрала "BLAST" в меню справа, затем нашла алгоритм "blastp", предназначенный для поиска участков сходства в последовательностях белков, и ввела в открывшееся окно идентификатор моего белка. Поскольку семейство (флавинредуктазы), которому принадлежит мой белок, довольно узкое, находок относительно немного (и их Query-cover весьма высок), я ввела дополнительное условие поиска - ограничила поиск таксоном Bacteria. Затем я получила 12 возможных гомологов, отобранных в соответствии со следующими параметрами:
Каждая находка имеет свои параметры, которые позволяют оценить её биологическую значимость:
- 1. Score - вес парного выравнивания исходной последовательности с найденной. Чаще всего используется матрица аминокислотных замен BLOSUM62. Этот параметр разделён на 2 колонки: Max Score - это максимальный вес, Total Score - это общий вес. Обычно Max score и Total Score совпадают. Если они отличаются, то находка выровнялась по нескольким участкам сходства.
- 2. Bit-score - нормализация посчитанного Score. Чем больше, тем лучше.
- 3. E-value (Expect) - это значение показывает, насколько случайно полученное выравнивание. Это количество находок с таким же или большим весом при поиске в базе данных случайных последовательностей. Например, если для находки e-value = 10, то это значит, что в базе данных случайных последовательностей найдётся 10 находок с таким же или большим весом. Чем меньше e-value, тем более статистически значима эта находка. i>
- 4. Query-cover - какой процент длины исходной последовательности выровнялся с находкой.
- 5. Ident - процент совпавших аминокислот.
- 6. Positives - процент похожих аминокислот (отмечается + в выравнивании).
- 7. Gaps - процент гэпов.
Таким образом, условными критериями гомологичности здесь для нас будут E-value<0.001 и Query cover не менее 70%.
При запросе ID Refseq BLAST определил суперсемейство данного белка, положение каталитических остатков в нём, домены:

Стоит отметить, что предположение о существовании данного белка было выдвинуто на основании данных о возможной минимальной длине гена, которая была определена неправильно. Поэтому и идентификатор был изменен с YP_007412963.1 на WP_003641675.1. По длине они полностью совпадают. Название старого белка - белок семейства нитроредуктаз (кодируется геном NC_020229.1), в то время как нового - NAD(P)H-зависимая хинонредуктаза (не указан ген и информации в целом существенно меньше, но добавлены еще функции). Проект ре-аннотации прокариотических геномов был обусловлен большим числом совпадающих последовательностей, присылаемых из разных лабораторий. Неохарактеризованным белкам теперь стали добавлять "WP"[1].
Также BLAST строит филогенетические деревья выровненных последовательностей:
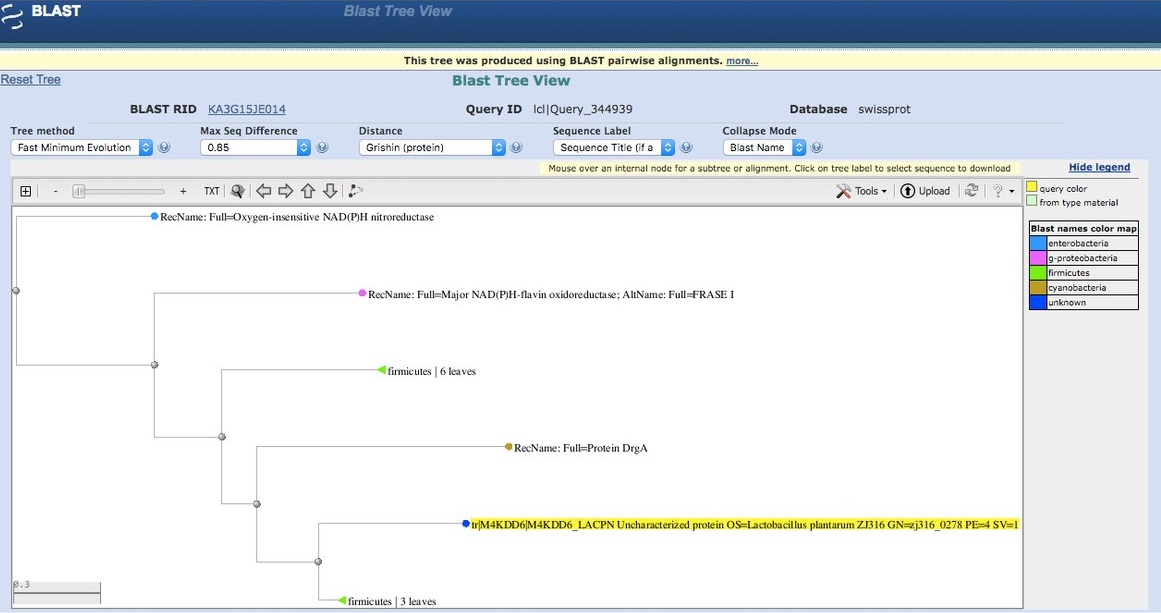
Древо, выданное программой BLAST, отражающее возможное родство. Поскольку, как было указано выше, данный белок только у бактерий (причем только а Грамм-отрицательных), дерево строится по этим семействам (Enterobacteria, g-proteobacteria, Firmicutes, Cyanobacteria).Однако, стоит отметить, что поскольку ограничений на поиск не вводилось, в выборку попали и 2 эукариотических организма, у которых нитроредуктаза имеет некоторую гомологию (довольно большое значение Е-value) с йодотирозиновой деионидазой. Поскольку наш белок принадлежит бактерии из семейства Firmicutes, здесь совпадений больше всего (в тоже время, и вариабельность в определенных участках, видимо, не играющих особой роли в функционировании белка) есть только у бактерийПо древу можно сказать, что В4LHB5_DROVI базальна по отношению к Е4XE80_OIKDI (имеют соответственно пролин и валин в 42). В других же верхних ветках валин заменяется на гомолог - лейцин или изолейцин. PAM250, neighbours
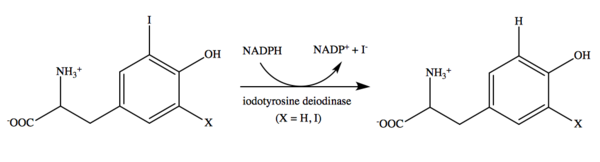
Реакция, которую осуществляет Iodotyrosine deiodinase, найденная у человека (sp|Q6PHW0.2|IYD1_HUMAN), свиньи Sus scrofa (sp|Q6TA49.1|IYD1_PIG), норвежской крысы (sp|Q5BK17.1|IYD1_RAT), орангутана Pongo abelii (sp|Q5REW1.1|IYD1_PONAB), Mus musculus (sp|Q9DCX8.1|IYD1_MOUSE)
| Находка | Длина выравнивания | Bit Score | E-value | Идентичные остатки,% | Сходные остатки, % | Выравнивание | Название белка |
|---|---|---|---|---|---|---|---|
| Лучшая | 217 | 454 | 4*10-162 | 100 | 58 | полученное BLAST | NAD(P)H-dependent quinone reductase |
| Из середины | 200 | 50.4 | 4*10-149 | 91 | 95 | полученное BLAST | NAD(P)H-dependent oxidoreductase |
| Худшая | 238 | 36.2 | 10 | 35 | 62 | полученное BLAST | Putative NAD(P)H nitroreductase SAS2409 |
Таким образом, гомологов, удовлетворяющих критериям - 12.
Затем было проведено множественное выравнивание с помощью программы muscle пакета EMBOSS. Как можно видеть далее, на N- и C-концах сохраняются невыровненные участки.

Также построено парное выравнивание худшей последовательности из гомологов и моего белка

Программа needle использует алгоритм Нидлмана-Вунша для построения глобального выравнивания двух последовательностей. Глобальное выравнивание подразумевает гомологию последовательностей по всей длине, туда включаются обе последовательности целиком. Сначала я получила парное выравнивание двух последних последовательностей в формате по умолчанию.
Затем, дописав опцию -aformat3 fasta я получила output файл в формате fasta
Далее я построила локальное выравнивание двух последовательностей - исходной и худшего и гомологов в формате fasta, используя для этого команду water на kodomo. Локальное выравнивание строится, когда в последовательностях есть негомологичные участки, тогда они исключаются и выравнивание идёт между гомологичными участками. Для получения локального выравнивания используют алгоритм Смита-Ватермана.
В следующем задании было предложено сравнить выравнивания. Как можно заметить, начальные участки (отсюда и длина) варьируют при разных алгоритмах. Так алгоритм Смита-Ватермана образает небольшой участок (есть в выравнивании бласта), однако сильнее различаются глобальные выравнивания muscle и needle. Вначале выравнивания участки не совпадают, потом я добавила несколько гэпов, чтобы выделить общие столбцы:

Скачать выравнивание выравниваний, проект
Скачать выравнивание выравниваний, FASTA
Посмотрим локальное и глобальное выравнивание негомологичных белков (с помощью needle и water). Вначале участки не совпадают, что обусловлено вырезанием при локальном выравнивании части несовпадающей последовательности. Соответственно, отсюда разный процент совпадений последовательностей.

Несовпадение последовательностей вначале можно заметить и на карте локального сходства.
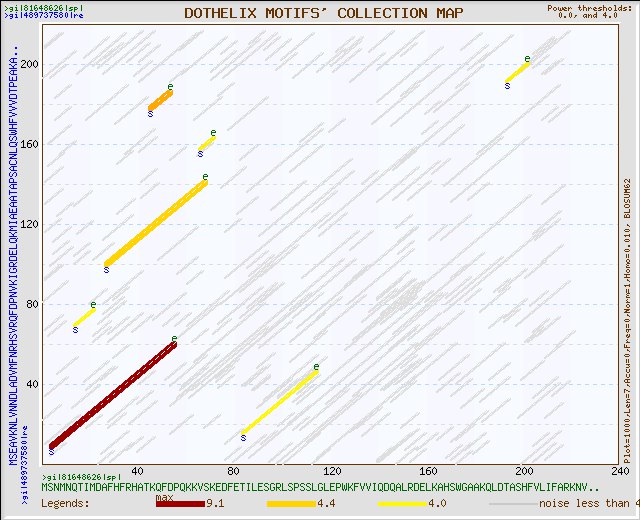
Затем было проведено парное выравнивание заведомо негомологичных белков - WP_003641675.1 и одного из цитохромов человека. Как и ожидалось, у них нет общих блоков. Локальное выравнивание показало большую схожесть, что неудивительно, учитывая, что данный механизм сравнивает участки, а не всю последовательность.
FASTA, выравнивание негомологичных последовательностей в программе needle
FASTA, выравнивание негомологичных последовательностей в программе water
[1] - Prokaryotic RefSeq Genome Re-annotation Project// URL: http://www.ncbi.nlm.nih.gov/refseq/about/prokaryotes/reannotation/
[2] - Introduction to Bioinformatics, 3rd Edition. Arthur M. Lesk