



Первая часть работы выполнялась Аксеновой Мариной
В первом задании требовалось, используя fuzznuc из пакета EMBOSS , найти все сайты, пересекающиеся с предсказанными сайтами транскрипционных факторов. Далее приведем описание программы fuzznuc и укажем опции, использовавшиеся при запуске. Были проверены 2 предсказанных мотива:
| Функция | Описание | Алгоритм |
| Поиск шаблонов в нуклеотидных последовательностях | Ищет специфичный PROSITE-style паттерн в нуклеотидных последовательностях. Такие паттерны являются характеристиками длиной последовательности (как правило, короткая). Они могут задать поиск точной последовательности или они могут допустить различные вариации, совпадения с варьирующей длиной последовательности и повторяющимися блоками в последовательности. Одна и более нуклеотидных последовательностей читаются с файла. Выходной файл - стандартный файл отчета EMBOSS, включающий такие данные, как расположение и скор (вес) всех совпадений. | fuzznuc разумно выбирает оптимальный алгоритм поиска |
*Далее сравнение проводилось при условиях, что на вход программы fuzznuc подавался геном бактерии, т.е. сайты метилирования искались в нем. 1.05.2017: видимо, так как, поиск fuzznuc ведется в upstream regions и указываются относительные координаты, в итоге мы можем соотносить координаты из выдачи FIMO (смотрим 1-4 находки) и выдачу fuzznuc - так мы далее будем оценивать пересечение сайтов метилтрасфераз и транскрипционного фактора. На вход программе fuzznuc подавался файл MT.pat, содержащий сайты связывания метилтрасфераз только нашей бактерии (для чистоты эксперимента), команда:
fuzznuc -sequence mot -pattern @MT-sites.pat -outfile fuzznucMOT.out
По 4 последовательностям я проверила паттерны, результаты можно увидеть в таблице 2.
| Название гена/локуса | Координаты мотива относительно гена | Координаты взятого участка | Длина взятого участка | Число найденных совпадений | Файл с мотивом | Файл с результатом |
| ydeW/STM4073 | 177..207 | 4283447..4283477 | 31 | 25 | mot4 | fuzznuc4.out |
| ego/STM4074 | 11..41 | 4283281..4283311 | 31 | 29 | mot1 | fuzznucMOT2.out |
Поскольку выдача fuzznuc по умолчанию ставит цепь "+", попробуем обратные цепи заменить на комплементарные с помощью Biopython: my_dna.complement()
Результат оказался аналогичен: скорее всего, метилазы (ну или fuzznuc) предпочитают положительные цепи.| Название гена/локуса | Координаты мотива относительно гена | Координаты взятого участка | Длина взятого участка | Число найденных совпадений | Файл с мотивом | Файл с результатом |
| ydeW/STM4073 | 215..235 | 4283281..4283311 | 31 | 25 | mot2 | fuzznucMOT2.out |
| ydeW/STM4073 | 49..79 | 4283447..4283477 | 31 | 20 | mot3 | fuzznucMOT3.out |
Таким образом, казалось бы, в таких коротких последовательностях не могло бы быть слишком много сайтов метилирования метилтрансферазами, однако как показывают результаты - регуляция транскрипции метилированием действительно очень распространена, во всяком случае у данного вида бактерий: всего сайтов, пересекающихся с сайтами (4) посадки транскрипционного фактор - целых 99.
Однако, как ни странно, поиск только лишь по сайтам нашей бактерии (скачаны с REBASE) ни к чему не привел. Поэтому было решено запустить с сайтами, выставленными на кодомо.
Для того, чтобы проверить, насколько сайты метилирования совпадают с сайтами связывания транскрипционного фактора, мы разбили задачу на несколько этапов:
1)ответить на вопрос, сколько сайтов совпадает по координатам, сколько сайтов перекрывается
2)ответить на вопрос, значимо ли отклоняются старты и концы двух типов сайтов метилирования
3)ответить на вопрос, можно ли утверждать, что концы и старты сайтов метилирования ниже
Для ответа на 2 и 3 вопросы был написан скрипт на R, на вход ему подавались координаты из выдачи FIMO.
Были проведены тесты Уилкокса - ответы на вопросы 3 и 2 соответственно:
Wilcoxon rank sum test with continuity correction data: met and fimo W = 31908, p-value = 1 alternative hypothesis: true location shift is less than 0
Здесь из р-value очевидно, что старт однозначно выше (однако картину слегка портят повторяющиеся значения).
Wilcoxon rank sum test with continuity correction data: met and fimo W = 31908, p-value = 4.396e-11 alternative hypothesis: true location shift is not equal to 0
То есть на уровне значимости 5% старт не отклоняется с вероятностью 4.396e-11 - отклоняется довольно сильно. Далее проделаем тоже самое для координат конца.
Wilcoxon rank sum test with continuity correction data: met and fimo W = 31272, p-value = 1 alternative hypothesis: true location shift is less than 0
Конец однозначно выше (p-value 1), однако картину слегка портят повторяющиеся значения.
Wilcoxon rank sum test with continuity correction data: met and fimo W = 31272, p-value = 7.126e-10 alternative hypothesis: true location shift is not equal to 0
То есть на уровне значимости 5% старт не отклоняется с вероятностью 7.126e-10 - отклоняется довольно сильно. Таким образом, старт и конец имеют довольно разные координаты. Говоря о пересечении данных - они есть, но не слишком часты - об этом косвенно свидетельствуют низкие значения p-value в тесте Уилкоксона с двусторонней альтернативой. Было замечено, что пересекаются сайты МТ и ТФ порой даже так, что сайты МТаз заходят друг на друга внутри ТФ, а за пределами upstream regions это происходит нечасто, что и логично, ибо лучший варинат регуляции активности - это регионы upstream, где еще нет смысловой последовательности самого гена.
У прокариот вообще метилирование не играет сильно большой роли в регуляции экспрессии генов (то есть, если сайт связывания ДНК-метилазы пересекается с промоторным или регуляторным участком, метилирование может оказывать влияние на связывание транскрипционных факторов [1]- в основном, метилирование используется для того, чтобы отличать "старую и новую" цепи ДНК. При транскрипции же здесь, возможно, вклад метилирования достаточно мал или нестабилен (может даже иногда происходит рекомбинация - так и регулируется). Таким образом, короткие сайты, например "GATC" скорее всего просто случайно совпали, а в базе данный сайт прописан как сайт метилирования в "вирусном иммунитете".
ЗАДАЧА: Используя REBASE проверьте есть ли в вашем геноме или геномах других штаммов того же вида ДНК метилтрансферазы с той же специфичностью. Укажите их в протоколе.
Итак, по запросу "Salmonella typhimurium LT2" в качестве организма, мы получаем таблицу, в которой указываются: номер организма в базе, латинское название вида (штамма), оптимальная температура роста, источник. Перейдя по ссылке с видом, мы получаем 14 находок, 10 из которых имеют некоторые паттерны, которые мы и будем проверять в задании 1. Поскольку каждая метилтрансфераза имеет свой сайт метилирования (но иногда они частично совпадают), далее число нахадок - это число совпадающих метилтрансфераз у разных штаммов.
Теперь проверим разные штаммы Salmonella typhimurium на совпадение паттернов.
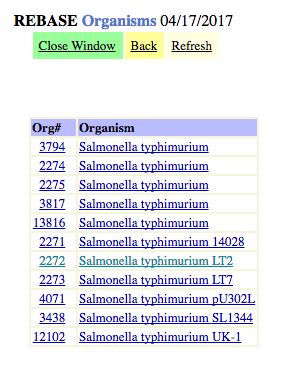
Рис. 2 Список находок по запросу Salmonella typhimurium
В распоряжении у нас 11 штаммов, включая наш. Далее будут указаны число находок, мотивы и соответствующие картинки.
Первая находка - только один сайт, совпадающим с первым из нашего штамма.
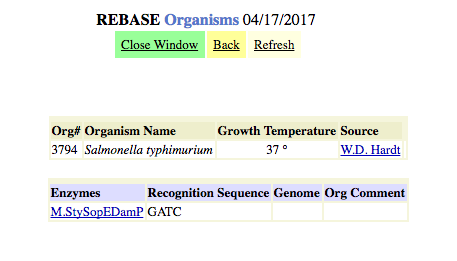
Рис. Список находок по запросу Salmonella typhimurium
Вторая находка - нет сайтов.
Третья находка - нет сайтов.
Четвертая находка - только один сайт, совпадающим с первым из нашего штамма.
Пятая находка - нет сайтов.
Шестая находка - 15 находок, совпадающих с нашим штаммом.
Седьмая находка - только один сайт, совпадающим с первым из нашего штамма.
Восьмая - наша.
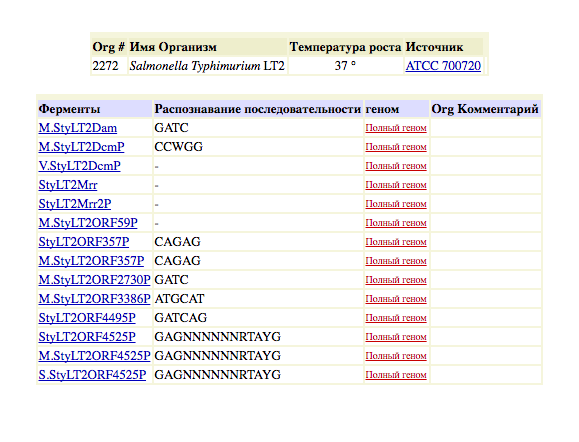
Рис. Список находок по запросу Salmonella typhimurium LT2
Девятая находка - 5 из 7 находок совпадают.
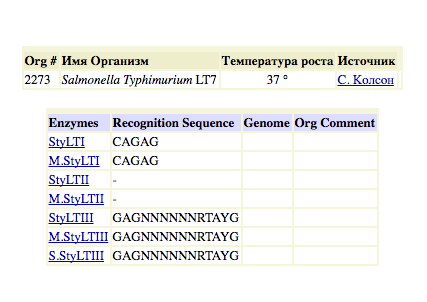
Рис. Список находок по запросу Salmonella typhimurium LT7
Десятая находка - только один сайт, совпадающим с первым из нашего штамма.
Одиннадцатая находка - только один сайт, совпадающим с первым из нашего штамма.

Рис. Список находок по запросу Salmonella typhimurium SL1344
Двенадцатая находка - нет сайтов, совпадающих с нашим штаммом.
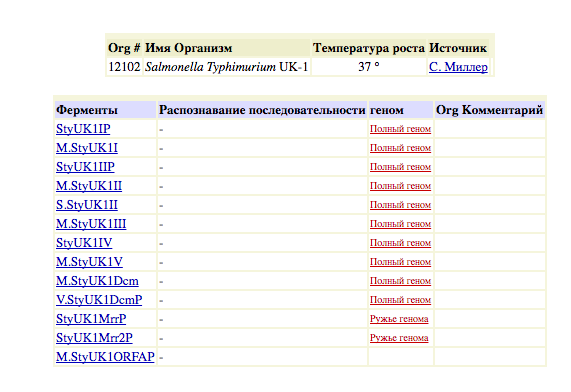
Рис. Список находок по запросу Salmonella typhimurium UK-1
Ссылки:
[1] NCBI DNA methylation in bacteria: from the methyl group to the methylome