



Множественное выравнивание — это выравнивание трёх и более последовательностей. Применяется для нахождения консервативных участков в наборе гомологичных последовательностей. В большинстве случаев построение множественного выравнивания — необходимый этап реконструкции филогенетических деревьев. Нахождение оптимального множественного выравнивания методом динамического программирования имеет слишком большую временную сложность, поэтому множественные выравнивания строятся на базе различных эвристик. Наиболее известные программы, осуществляющие множественное выравнивание — Clustal (http://www2.ebi.ac.uk/clustalw/), T-COFFEE (англ.) (http://tcoffee.vital-it.ch/cgi-bin/Tcoffee/tcoffee_cgi/index.cgi), MUSCLE (англ.) (http://www.drive5.com/muscle/) и MAFFT (англ.) (http://mafft.cbrc.jp/alignment/software/). Имеются также программы для просмотра и редактирования множественных выравниваний, например Jalview (англ.) или русскоязычный UGENE [1].
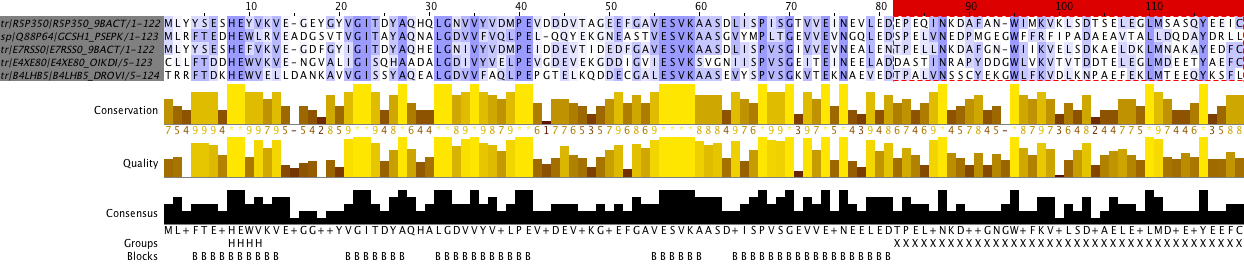
В качестве блока были выбраны те участки, в которых 1)наблюдались абсолютно консервативные позиции, 2)гомологичные остатки (то есть кодирующиеся из "родственных кодонов" - от предкового произошло не более 2 замен) и 3)функционально консервативные (попадают в одну группу, по списку, предложенному на сайте), кроме того, внутри блока не должно быть гэпов. Таким образом, получаем 17 блоков. Учитывая, что данные белки принадлежат к одному семейству, это информативный и нормальный результат. Блоки отмечены символом "В" в добавленной строке "Bloks". Затем объединим в группу "Н" участок 8-11, где все позиции абсолютно консервативны у двух нижних (на рисунке) последовательностей. Как можно заметить, в отдельных положениях Н-участков между конкретными последовательностями также наблюдается гомология, что может быть обусловлено рядом причин. Во-первых, это можно объяснить через горизонтальный перенос генов (трансдукция, трансформация, коньюгация), так как все обладатели данных белков - бактерии. Данное объяснение, однако, не согласуется с тем, что такие участки единичны, больше похожи на точечные мутации, чем на ГПГ. Другое возможное объяснение - данные участки расположены в поверхностных участках белка или в "хвостах", т.е. так называемых вариабельных участках (активный центр, лиганд расположены в других частях молекулы, ключевые остатки, впрочем, тоже). Найдем в PyMol эти последовательности - действительно, они не входят в гидрофобное ядро и,видимо, не вносят существенный вклад в функционирование белка. Кроме того, наличие общих остатков у определенных белков может свидетельствовать в пользу их родства (или даже родства их обладателей), большего чем с другими белков из выбранных (в чем можно убедиться, построев древо посредством ). Наименее вероятное объяснение - в настоящий момент эти участки лишь реликты когда-то консервативных, ставших вариабельными вследствие различных мутационных событий. Другой пример - консервативные аминокислоты близ положения 42.
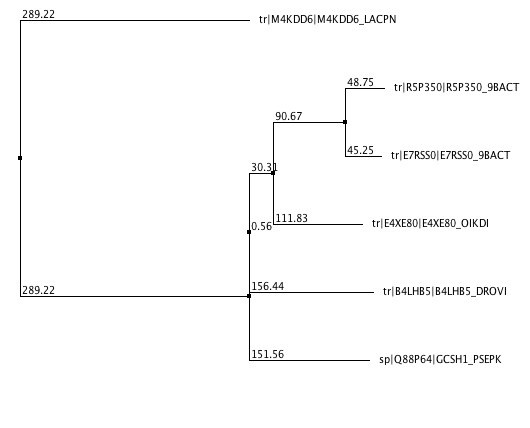
Древо, отражающее возможное родство. Здесь мы уточняем вариабельность в положении 42. По древу можно сказать, что В4LHB5_DROVI базальна по отношению к Е4XE80_OIKDI (имеют соответственно пролин и валин в 42). В других же верхних ветках валин заменяется на гомолог - лейцин или изолейцин. PAM250, neighbours
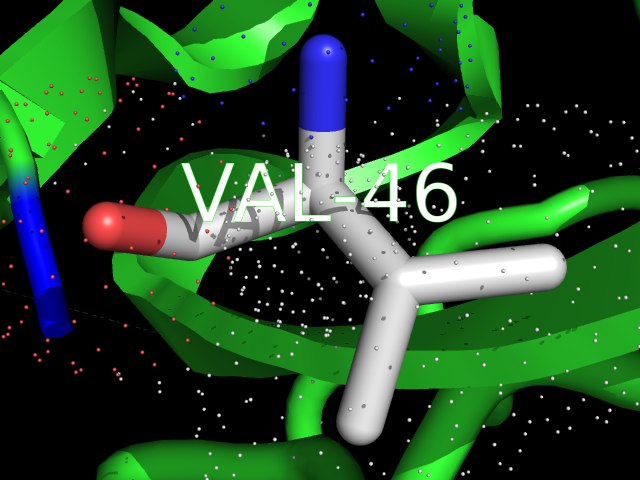
Изображение остатка 42, в гомологе, для которого известна структура, в позиции 46 (было установлено при добавлении в выравнивание). Получено в PyMol
Для подсчета консервативных позиций я выбрала блок 1 (4-14). Абсолютно консервативная позиция - это такая колонка, в которой во всех выравниваниях стоит одинаковая аминокислота. В этом блоке их 11, в процентном соотношении 18,18%. Абсолютно функционально консервативная позиция - это такая колонка, в которой во всех выравниваниях стоят аминокислоты одной и той же группы. Таких колонок 6, их доля в процентах - 54,55%
Матрица BLOSUM основана на базе данных выровненных последовательностей белков BLOCKS - BLOcks SUbstitution Matrix. Из семейства родственных белков, выравниваемых без пропусков, Хэникофф и Хэникофф вычислили отношение числа рассматриваемых пар аминокислотных остатков на любой позиции, к числу пар, ожидаемых для всех аминокислотных последовательностей. Результат представлен в виде логарифмов частот замен. Чтобы избежать излишнего влияния родственных последовательностей, Хеникофф заменили группы белков, которые имеют идентичность поеследовательности выше, чем поороговая, либо одним представителем, либо их средневзвешенным значением. Порог в 62% создает часто используемую матрицу замещений BLOSUM62 [2].
Для построения LOGO для блока 2 мной был использован сервис WebLogo. Предварительно с помощью программы cons EMBOSS была получена консенсусная последовательность.
LOGO - это наглядное графическое представление множественного выравнивания. Величина буквы в каждой позиции на рисунке говорит о консервативности аминокислоты в этом положении.По умолчанию аминокислотные последовательности окрашиваются по гидрофобности. Гидрофобные аминокислоты окрашены черным, гидрофильные синим, нейтральные зеленым.На основании данных лого или визуальным анализом составим паттерн блока 2:
<[VI]-G-I-[TS]-[DAQS]-[YH]-A>
Необходимо отметить, что видимо остаток положительно заряженного гистидина и отрицательного глутамата, находясь рядом (связаны пептидной связью, не позволяющей вращение), не взаимодействуют между собой, зато образуют солевые мостики с другими остатками (пихнуть изображение), видимо, их присутствие обязательно (абсолютная консервативность) для осуществления функции белка. Также отсутствие участков делеций или инсерций здесь подтверждает консервативность
Теперь добавим заведомо негомологичную последовательность - белок YP_007412963.1. Затем добавим 3 гэпа - в местах, где хотя бы в одной из последовательностей есть гэп. Длина значительно превосходит длину наших последовательностей, поэтому обрежем до одинаковой. Теперь совпадений в остатках стало чуть больше. Приведем список совпадений в блоках и с частотами встречаемости:

Таким образом, процент совпадений рассчитаем как произведение (независимые события) -0,152% -, что неудивительно, ибо выбранный белок негомологичен данным
Самый длинный участок между блоками обозначим за "Х". В нем всего 39 позиций и лишь 1 гэп (надо сказать, в целом во всех позициях лишь 3 гэпа, что абсолютно нормально, ибо белки гомологичны), таким образом их (окружающие блоки + сам участок)
Как можно видеть на рисунке, мне не удалось найти хоть сколько нибудь удачный блок "выравнивания", по которому можно было бы судить о гомологии. В "блоках" (так мы назовем участки из менее 3 колонок, где есть хоть какое-то совпадение) практически нет совпадающих аминокислот, а также много гэпов. По такому "выравниванию" сразу можно заключить, что белки негомологичны. Здесь длина была обрезана до одинаковой. Здесь нет абсолютно консервативных колонок, выделены лишь те, что могли бы говорить о гомологии, если бы входили в реальный блок (таких 5 колонок из 818 (0,61%)).

Использованные последовательности:
[1] - Wikipedia Sequence alignment// URL: https://en.wikipedia.org/wiki/Sequence_alignment
[2] - Introduction to Bioinformatics, 3rd Edition. Arthur M. Lesk