



Шаги, выполненые при запуске программы retree пакета PHYLIP (использовалась локальная версия)
1.Скопировать содержимое файла *.nwk в файл intree, находящийся в той же дирректории, что и файл запуска программы retree.
2.Вызвать программу.
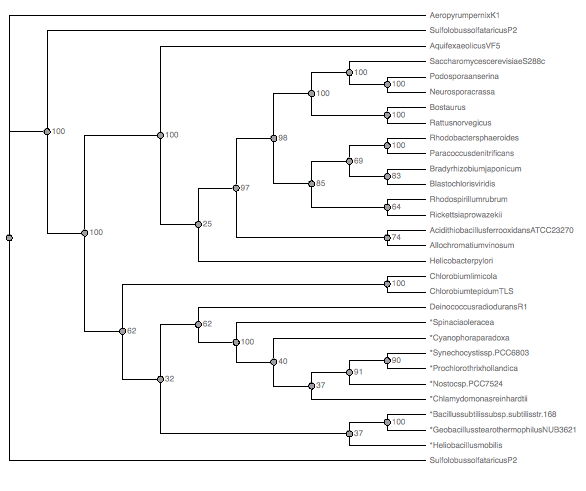
3.Указать "M" - укоренение в среднюю точку (то есть был найден самый "долгий" путь между таксонами, и на нем взята середина). Укоренение создало 2 ветви - {ENTFA, STRPN, STRP1, LACLM} vs {LACDA, LACAC, CLOTE}.
4.Визуализировать в програмее MEGA7.
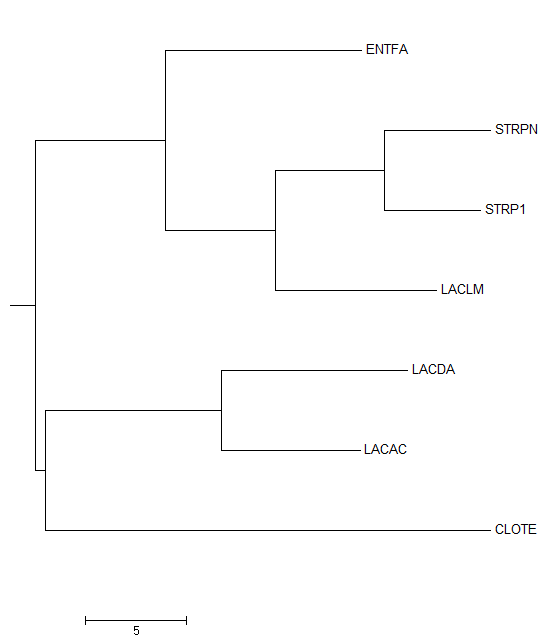 | 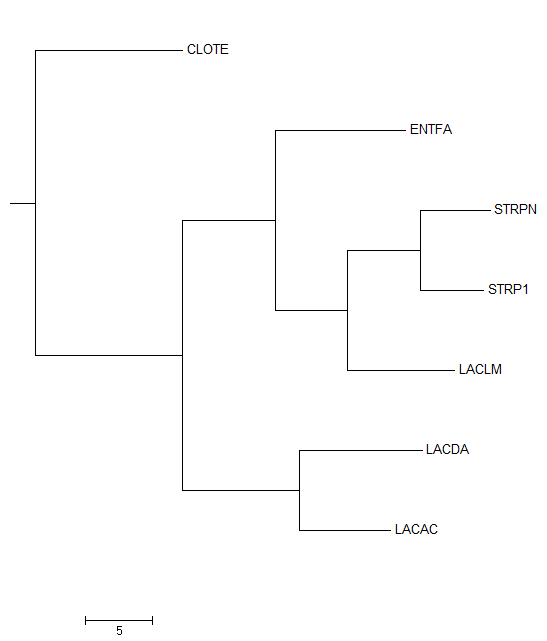 |
Как можно заметить, ветвь {LACDA,LACAC,CLOTE} содержит 2 представителя класса лактобацилл, в то время как CLOTE - единственная клостридия, кроме того LACLM также лактобацилла. Таким образом, данное древо неубедительно - единый таксон довольно близких организмов - лактобацилл - разделился по 2 ветвям. В дополнение : CLOTE базален в соответствии с древом видов по отношению ко всем выбранным организмам, а в случае переукоренения только по отношению к ветви {LACDA,LACAC}.
В статье [1] для укоренения древа используется внешняя группа - археи. Вообще обычно археи выбираются "эталоном для укоренени" в случаях, если все другие организмы не являются прокариотами или если мы очень уверены, что данный белок архей действительно древнее бактериального (кстати, позже в статье все же говорится о том, что гомологии с археями по cyt-bc не обнаружилось). По сути, нам нужно проверить, имели ли они право укоренить древо подобным образом. Они использовали алгоритм выравнивания CLUSTAL с матрицей BLOSSUM и по некой причине они не обосновали это. Итак, я с помощью команды (приводим далее выравнивание предположительных белков)
seqret art2 phylip::aln.phylip
перевела в формат PHYLIP, а затем запустила программу seqboot и получила число выравниваний бутстрэпа, в 10 раз меньшее, чем в статье - 5000 реплик - это довольно много и ненужно, только тормозитт работу. Теперь нужно разделить файл с выравниваниями на отдельные - скрипт. Затем с помощью несложных команд bash я получила дерерья,построенные на основании этих последовательностей. После этого я использовала программу consense, подавая на вход файл с деревьями - так я получила консенсусное древо - оно нам потребуется для проверки наличия горизонтального переноса генов в данном случае. Мы это сейчас (в рамках практикума) проверять не будем - нужно не по белкам, а по рРНК. *Хотя в замечательной программе UGENE все можно было сделать лишь движением руки.
*TREX может примерно сказать, есть ГПГ или нет: на вход нужно подать дерево видов и дерево по генам.
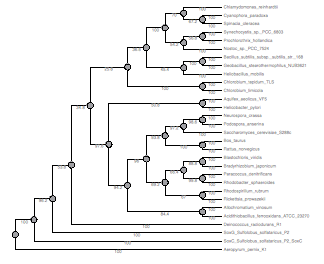 Консенсусное древо
Консенсусное древо
Теперь вернемся "с небес на землю" и построим древо методом neighbor-joining (матрица Kimuro) с 5000 репликами (но можно и 500). Также были построены деревья методами ML, RAxMLи MrBayes.
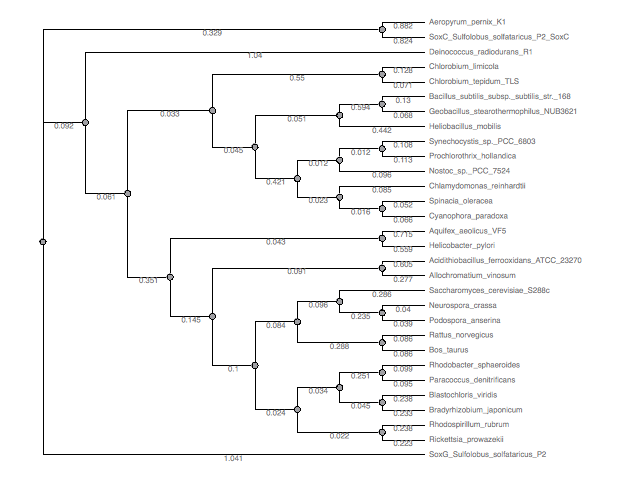
Даже и без укоренения в среднюю точку очевидно, что археи образуют единую кладу, а Deinococcus radiodurans базален по отношению по всем организмам.
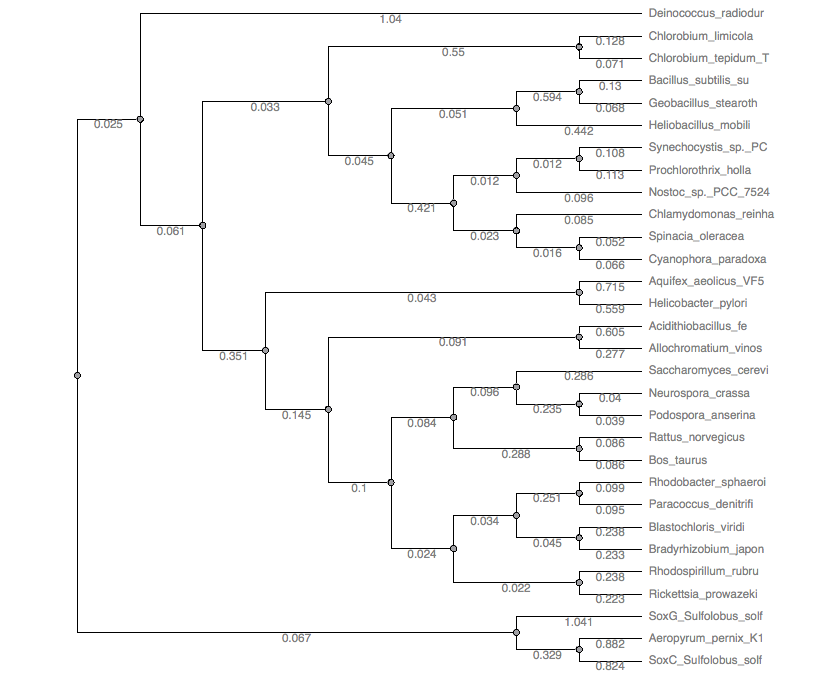
А затем PhyML:
Как можно заметить, Deinococcus radiodurans не занимает базального положения по отношению к другим таксонам, ветвь с поддержкой 0.184 является здесь объединением нескольких ветвей древа из статьи, Aquifex aeolicus имеет иное расположение.
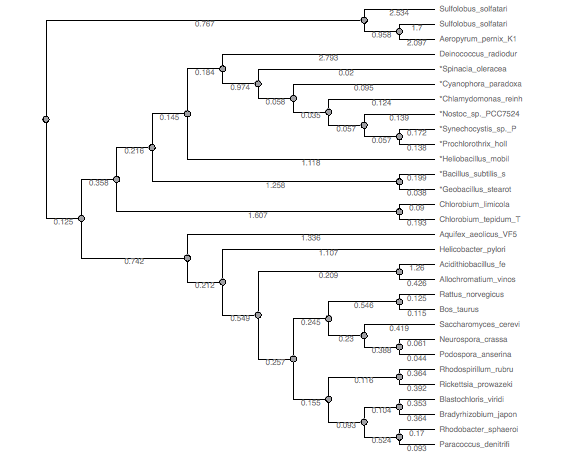
И RAxML:
Археи здесь разделились, но теперь Deinococcus radiodurans базален по отношению к цианобактериям, четко видно разделение на 2 группы (2ая включает грибы и некоторые бактерии). Таким образом, здесь наиболее явный кандидат на "корень" - Aquifex aollius, кроме того в одном из деревьев выше он был "где-то рядом" с Deinococcus radiodurans.
И MrBayes:
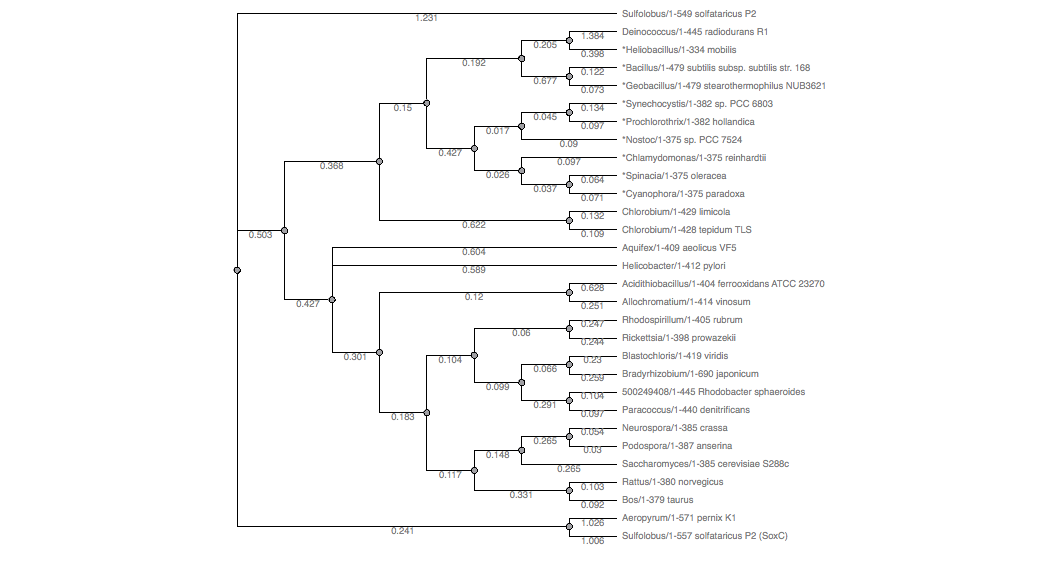
Здесь Deinococcus radiodurans не базален по отношению ко всем, а находится в одной кладе с Geobacillus, Heliobacillus, Bacillus - это ветвь базальна по отношению к цианобактериям. Ветви Aquifex и Helicobacter имеют довольно низкую поддержку (по NCBI они и не являются представителями общих таксонов).
На основании полученных данных, я бы отметила, какие ветви отделены наиболее - это археи (2 верхние, 1 нижняя), затем я бы их убрала (так как слабая гомология) и искала бы "новый корень" среди самой базальной ветки - это Deinococcus radiodurans (или Aquifex aollius, в идеале надо-бы взять много белков и построить объединенное древо). Таким образом, выбор архей в качестве аут-группы вполне обоснован: на всех деревьях есть эта нетривиальная ветвь. С другой стороны, есть данные в пользу базальности Deinococcus radiodurans, в который также можно переукоренять древо. По топологии с деревом из статьи совпали - это только NJ (1 из 4 использованных), так что я бы не стала доверять авторам. Кроме того, они использовали простейшую матрицу замен, которая, вероятно, не учитывает сложности модели в данном случае.
**20 марта
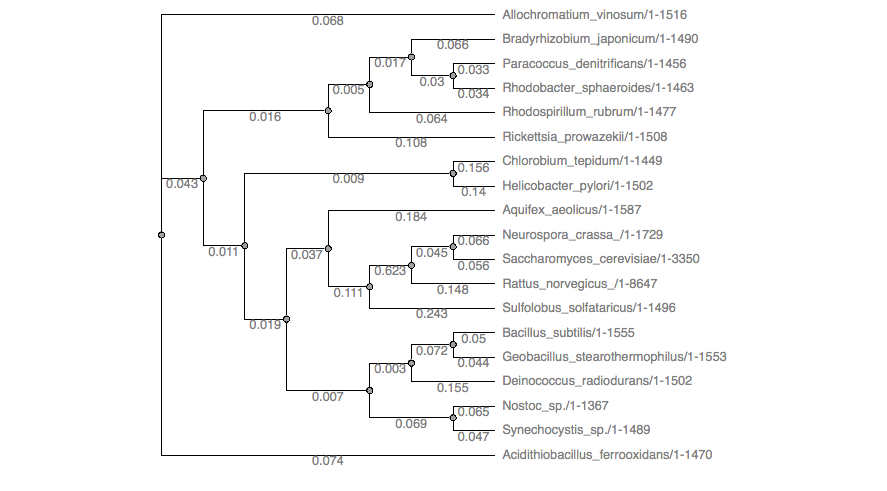
Мне все же захотелось довести дело до конца и я решила составить дерево по 16S рРНК, для чистоты эксперимента я брала рРНК тех же штаммов, если они были. Поскольку во многих файлах не встречается по несколько генов рРНК, я решила, что составлять объединенное древо здесь не уместно. Для эукариот я использовала BLAST, так как число файлов .frn слишком большое. Некоторые последовательности не удалось найти (Podospora anserina), возможно, по причине ошибки секвенирования/реаннотации, так что на этом этапе результаты из статьи вызывают сомнения. Поскольку 2 гриба у нас уже есть, проигнорируем этот "незначительный" факт. В файле с Aeropyrum pernix K1 вообще нет 16s.
Итак, я получила выравнивание Clustal (такое же и было в статье) и построила древо методом neighbor-joining. Как можно видеть, оно немного отличается от белковых. Ветвь 0.037 действительно наталкивает на мысль о ГПГ, так как неожиданно (как и в белковых) бактерии и высшие эукариоты оказываются вместе (интересно, что сервер T-rex выдал результаты о ГПГ). Так что хотя бы здесь автор статьи не соврал. *Здесь для сравнения дерева с рРНКовым использовалось "консенсусное" из начала этого задания.
 Ссылки:
Ссылки:
1.Schutz M, Brugna M, Lebrun E, Baymann F, Huber R, Stetter KO, Hauska G, Toci R, Lemesle-Meunier D, Tron P, Schmidt C, Nitschke W. Early evolution of cytochrome bc complexes. J Mol Biol. 2000;300:663–675.