Я получила последовательности с расширением *.frn в соответствующих директориях: я проверила штаммы, у которых я брала белки в предыдущем практикуме в Uniprot - только нужного штамма ENTFA не оказалось, поэтому я взяла следующий в списке.
В каждом файле *.frn находится как минимум два гена, кодирующих 16S рРНК, помимо этого здесь представлены все гены, кодирующие другие виды РНК. Я извлекла только 16s рРНК, затем объединила все файлы *.fasta командой cat file1 file2 > file3. Полученные последовательности были выровненны программой Mafft (она подходит для выравнивания нуклеотидных последовательностей, в то время как Muscle - нет). Из выравнивания стало понятно, что так как последовательности почти идентичны, скорее всего это слегка мутировавшие копии соответствующих генов рРНК (что подтверждается, скорее всего и тем, что генов по 2 для каждой 16S). Дальше есть несколько путей построить древо:
1.Можно взять 2 последовательности, так как это минимальное число генов 16s рРНК в файле - возьмем по два гена из каждого организма и построим независимо выравнивания, а затем составим консенсус. Однако, стоит отметить, что если даже мы и выбираем последовательности схожей длины, учитывая, что они могт отличаться всего несколькими буквами, мы берем во внимание факт того, что для таких очень консервативных последовательностях будет сложно установить возможный ход эволюции - мы можем просто не взять ортологи и построить 2 типа деревьев по неортологичным генам. Поэтому этот путь должен привести нас к перебору последовательностей и поиску лучшего выравнивания, чего делать не будем.
2.Объединим все выровненные последовательности в 1 файл, расположим так что "под одним организмом располагаются все его гены" , а затем удалим мнемоники всех генов одного организма, кроме первой (Jalview "сошьет их в один белок") - в данном случае так можно делать, так как последовтельности почти не отличаются, гэпов не много. Затем "подрежем" плохо выравненные концы программой trimAL:
./trimal -gt 0.5 -st 0.001 -w 1 -cons 5 -in concat_al.mfa > con_clear.mfa
То есть иначе говоря:
Пусть в записи каждого штамма есть некоторое число последовательностей нужной нам РНК, причем в записях разных штаммов, как правило, это число разное. Тогда будем извлекать последовательности примерно одинаковой длины (у разных штаммов). Затем сделаем парное выравнивание последовательностей у каждого штамма, а затем построим множественное выравнивание, по всем парным. На этом этапе можно удалить идентификаторы и как бы "слить последовательности одного штамма в одну" (что как раз породит множество ошибок, так как фактически далее мы будем анализировать в принципе не связанные между собой эволюцией последовательности (даже по сути не отдельные другие гены, а лишь копии, в которых есть, скорее всего, случайные мутации)).
*Опции помогают нам подбирать длину, которую мы бы хотели отрезать.
Затем для достоверности лучше построить реконструкции разными методами, что отражено в таблице.
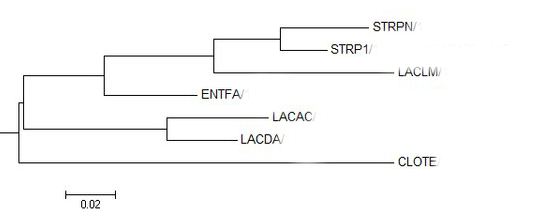 Рис.1 Maximum likelyhood Рис.1 Maximum likelyhood |
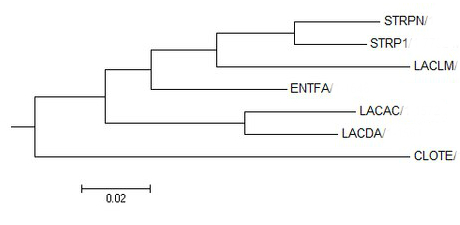 Рис.2 Neighbor-joining Рис.2 Neighbor-joining |
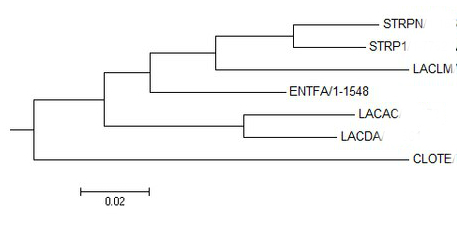 Рис.3 Minimum evolution Рис.3 Minimum evolution |
 Рис.4 MrBayes Рис.4 MrBayes |
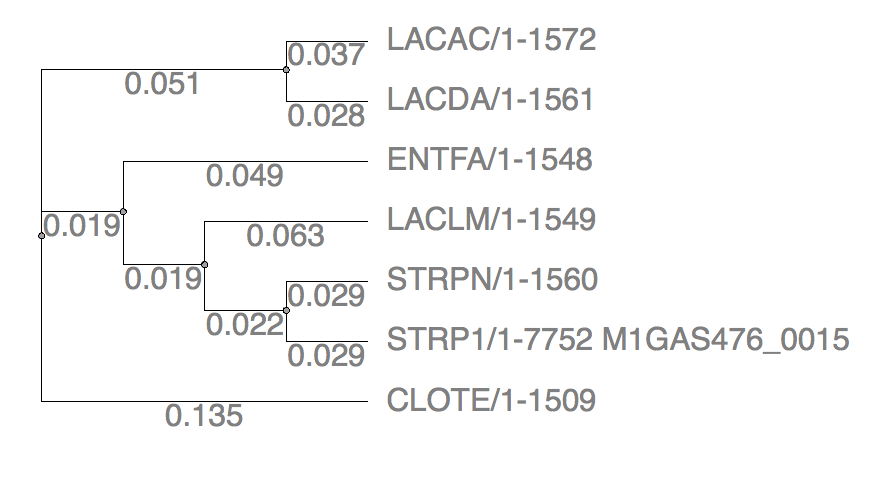 Рис.5 Neghbor-joining by PHYLIP Рис.5 Neghbor-joining by PHYLIP | |
 Рис.6 Maximum likelyhood by PHYLIP Рис.6 Maximum likelyhood by PHYLIP |
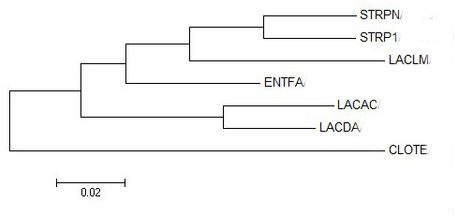 Рис.1a Maximum likelyhood Рис.1a Maximum likelyhood |
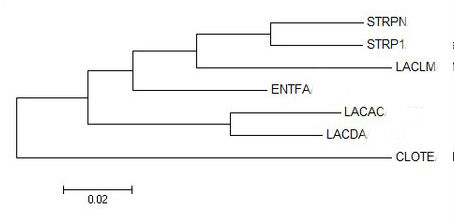 Рис.2a Neighbor-joining Рис.2a Neighbor-joining |
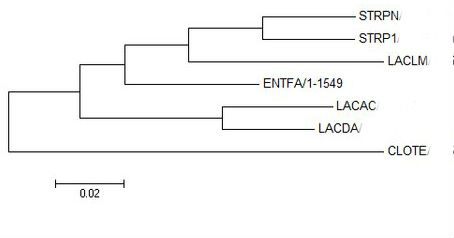 Рис.3a Minimum evolution Рис.3a Minimum evolution |
 Рис.4a MrBayes Рис.4a MrBayes |
 Рис.5a Neghbor-joining by PHYLIP Рис.5a Neghbor-joining by PHYLIP |
 Рис.6a Maximum likelyhood by PHYLIP Рис.6a Maximum likelyhood by PHYLIP | |
| Осуществление способа 1, далее указаны тэги локуса:
Для первого выравнивания CLOTE = CTC0r03 ENTFA = HMPREF0351_r10001 LACAC = LBA2001 LACDA = Ldb0054 LACLM = llmg_rRNA_1 STRPN = SP_rrnaA16S STRP1 = M1GAS476_0015 Для второго выравнивания CLOTE = CTC0r18 ENTFA = HMPREF0351_r10004 LACAC = LBA2012 LACDA = Ldb0798 LACLM = llmg_rRNA_48a STRPN = SP_rrnaD16S STRP1 = M1GAS476_0095 | |
Таблица 2. Осуществление способа 2
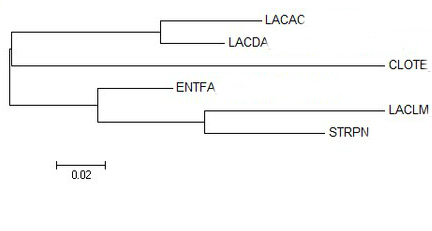 Рис.7 Maximum likelyhood Рис.7 Maximum likelyhood |
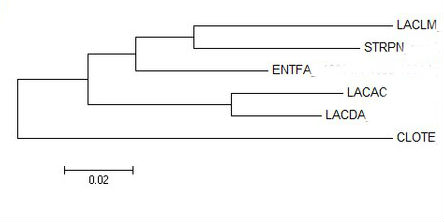 Рис.8 Neighbor-joining Рис.8 Neighbor-joining |
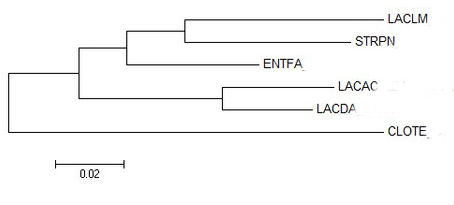 Рис.9 Minimum evolution Рис.9 Minimum evolution |
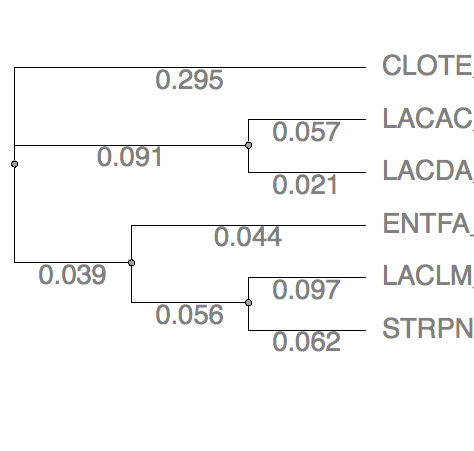 Рис.10 MrBayes Рис.10 MrBayes |
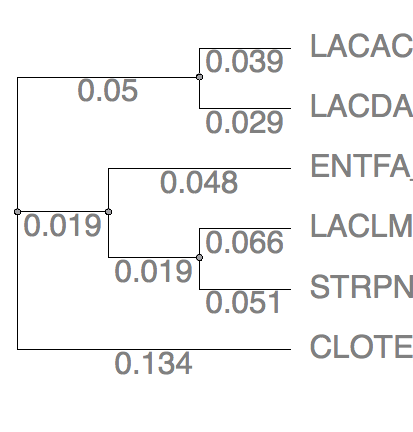 Рис.11 Neghbor-joining by PHYLIP Рис.11 Neghbor-joining by PHYLIP |
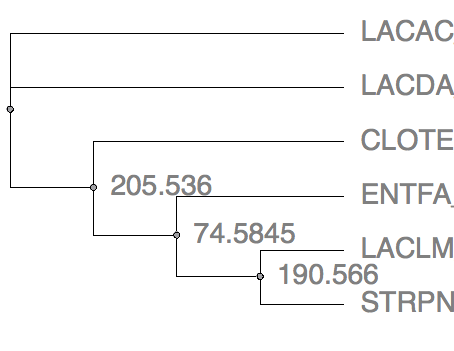 Рис.12 Maximum likelyhood by PHYLIP Рис.12 Maximum likelyhood by PHYLIP |
Как видно из абсолютно всех реконструкций, топология абсолютно всех ветвей в целом совпадает с эталонным древом (варьирует только поддержка в зависимости от метода).
*RAxML выдал неудовлетворительные результаты, поэтому здесь их не будет.Из-за того, что последовательности каждого варианта генов 16S выравнивались сначала парным выравниванием (уже порождаются гэпы, а из-за того, что последовательности почти идентичны, штраф больше), а затем множественным относительно друг друга, видимо, появлялись довольно большие штрафы за гэпы, поэтому перечисленные два способа, вероятно, не являются оптимальными. Поэтому далее я сделала реконструкции филогении указанными выше методами для набора из генов 16S рРНК, где для каждого организма представлен только один ген. Кроме того, вряд ли мы сможем применить программу trimal - если гэпов в выравнивании много, то у нас обрежется довольно много участков.
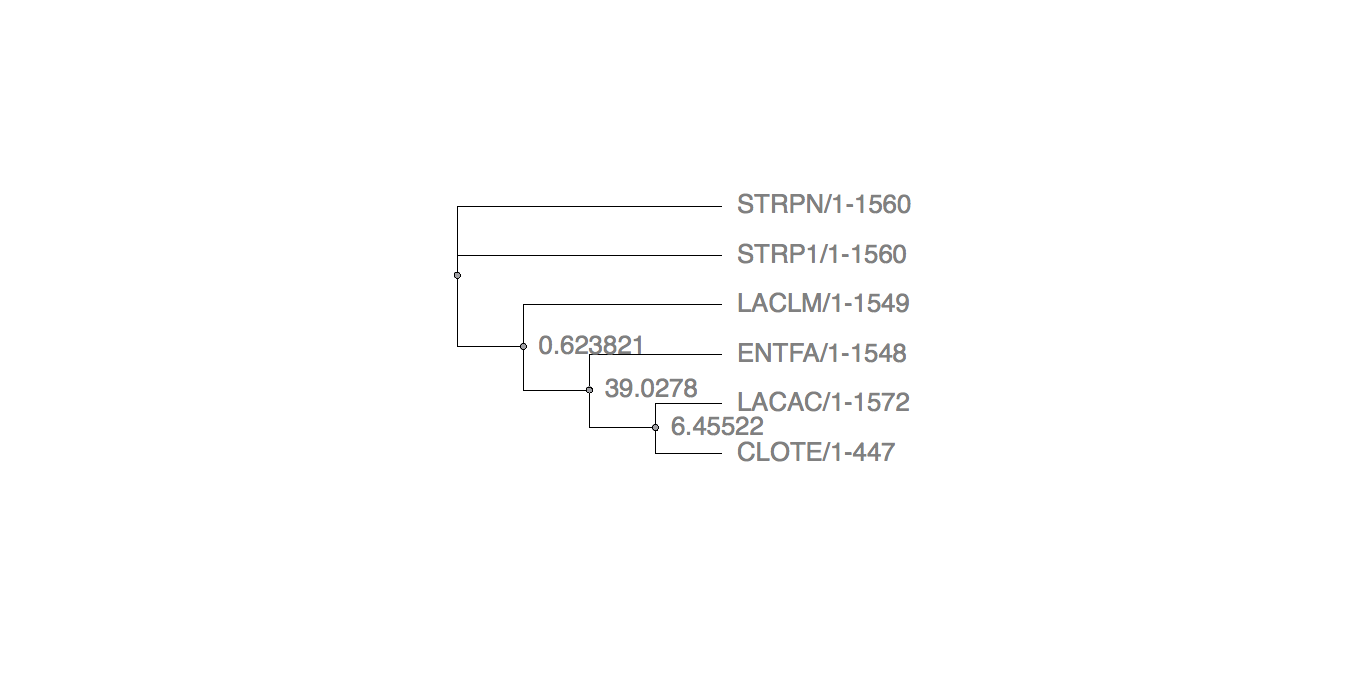 Рис.13 Maximum likelyhood (PhyML) Рис.13 Maximum likelyhood (PhyML) |
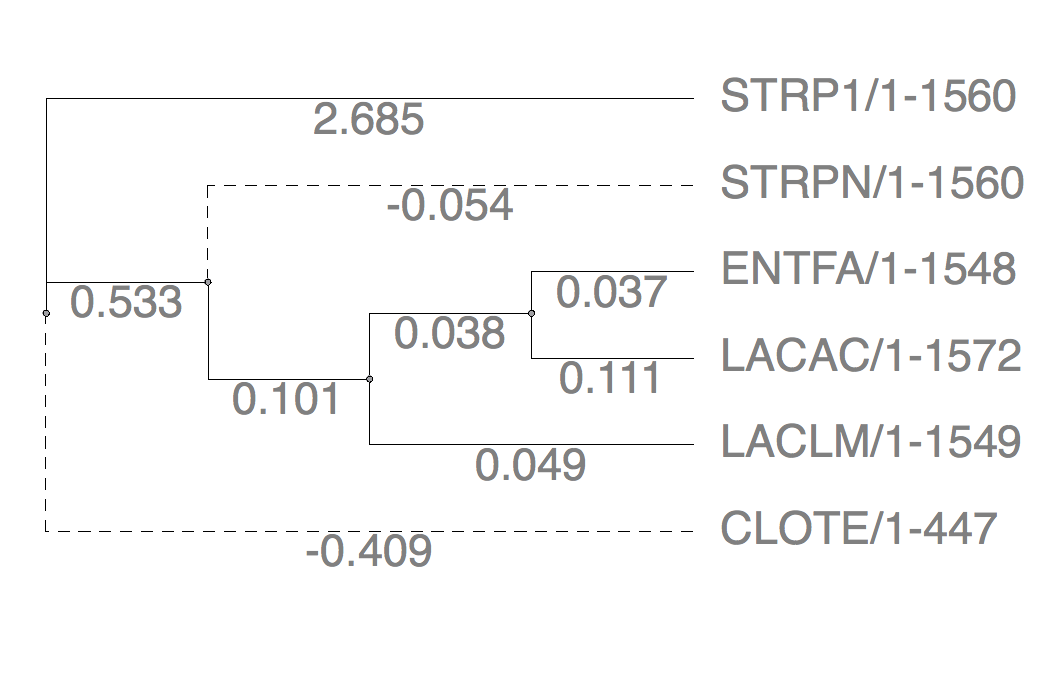 Рис.14 Neighbor-joining Рис.14 Neighbor-joining |
 Рис.15 MrBayes Рис.15 MrBayes |
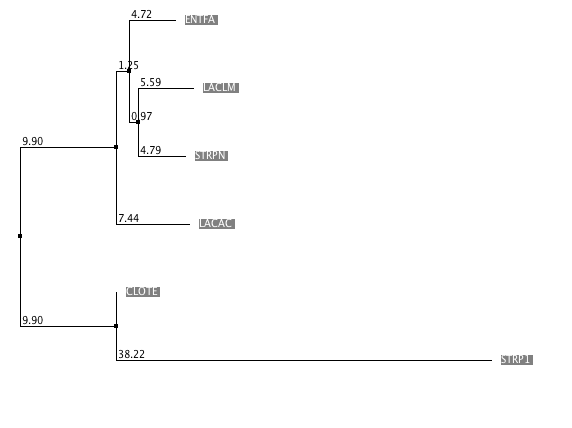 Рис.16 Neighbor Joining Using % Identity Рис.16 Neighbor Joining Using % Identity |
Древо, построенное методом PhyML наиболее точно отражает по топологии "эталонное". Neighbor-joining показал весьма недостоверные результаты, так как есть ветви с отрицательной поддержкой и топология отличается от "эталона". Пожалуй, единственное отличие этих реконструкций от эталона - группировка CLOTE и STRP1 в одну кладу.Таким образом, скорее всего все же более достоверными являются реконструкции на основе белков: 4 параметра для матриц замен не позволяют нам сказать, была ли замена в итоге синонимичной или нет или предпочтение зпмен определенных остатков.
Поскольку я неудачница и оставила в пятницу флешку в кабинете биоинформатики, а мега у меня на ней, придется пока что (на момент выходным) построить реконструкцию филогении другими программами.
Сначала я искала гомологи с помощью blastp с командами:makeblastdb -in infile -dbtype prot -out out_db - так создается база для поиска blastp -query bacsu -db clote_db -out clote.blast - этой командой я нашла гомологи в clote заданног белка.
По запросу в протеомах
>clote 8 находок
>lacac-6
>lacda-6
>entfa-7
>laclc-2
>strp1-5
>strpn-5
*Отбирались находки, e-value которых меньше 0,001
Затем были построены реконструкции филогении все теми же методами
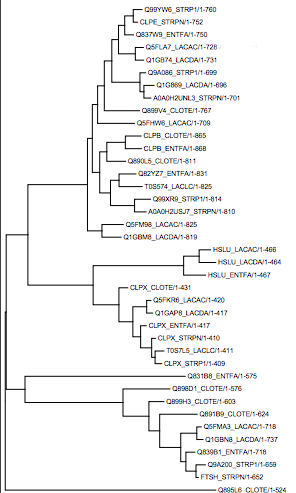 Рис.13 Maximum likelyhood Рис.13 Maximum likelyhood |
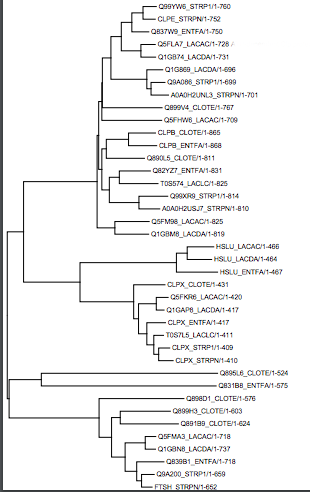 Рис.14 Neighbor-joining Рис.14 Neighbor-joining |
 Рис.15 Minimum evolution Рис.15 Minimum evolution |
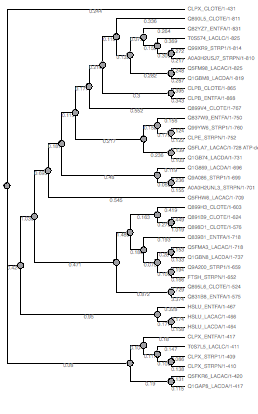 Рис.16 MrBayes Рис.16 MrBayes |
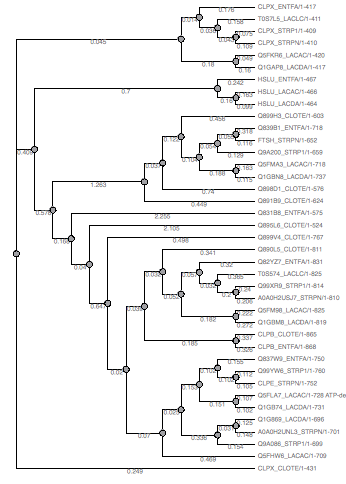 Рис.17 Neghbor-joining by PHYLIP Рис.17 Neghbor-joining by PHYLIP |
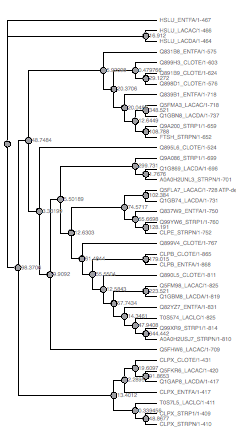 Рис.18 Maximum likelyhood by PHYLIP Рис.18 Maximum likelyhood by PHYLIP |
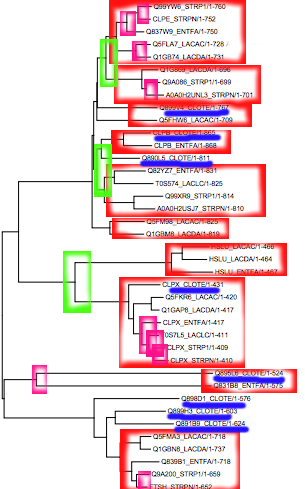
Теперь привожу древо, в котором группы белков уже выделены (это NJ из таблицы). Светло-зеленым отмечены дупликации, подтверждается тем, что два белка выполняют разные функции и просто являются разными, а присутствуют практически у все - белки CLPE, CLPX и HSLU представляют собой разные субъединицы АТФ-зависимой протеазы. Интересно, что CLOTE, по-видимому имеет еще одну субъединицу - о чем показвает его положение в нескольких ветвях. Фиолетовые рамки показывают видообразование - то есть показывают появление ортологов. Паралоги отмечены синим подчеркиванием (под паралогами понимаются два гомологичных белка из одного организма (выделены все гомологи из организма CLOTE, попарно являющиеся друг другу паралогами).Красные рамки - ортологичные группы. *Вообще для меня стало неожиданностью, что результаты абсолютно по всем реконструкциям разными методами абсолютно совпали - значит, мы прямо-таки почти однозначно можем установить ортологи и паралоги.