Алгоритм BLAST используется для поиска гомологичных последовательностей. Существуют отдельные вариации алгоритма для различных запросов: blastp (поиск аминокислотных последовательностей), blastn (поиск по нуклеотидным последовательностям), blastx (поиск в аминокислотных базах данных по нуклеотидному запросу) и так далее. В этом практикуме используется первый из перечисленных вариантов. Работать с программой можно на этом сайте.
Задачей был поиск гомологов в базе данных UniProtKB/Swiss-Prot для последовательности белка из первого семестра – калий-связывающего белка из семейства Ktr (KtrA). Его последовательность в формате fasta можно скачать тут.
Характеристики одной из находок представлены в таблице 1.
Таблица 1. Сведения о полученных выравниваниях
| Description | Max score | Total score | Query cover | E value | Ident | Accession |
|---|---|---|---|---|---|---|
| RecName: Full=Ktr system potassium uptake protein C; Short=K(+)-uptake protein KtrC; AltName: Full=ORF4 | 248 | 248 | 95% | 3e-81 | 56% | P39760.1 |
В колонке Description содержится описание белка, соответствующего найденной последовательности. Одна находка может содержать несколько участков сходства, тогда, максимальный вес (Max score) их этих участков будет не равен сумме этих весов (Total score). Если же наложение одно – они равны, как в случае выбранной находки. Указаны нормированные веса. То, какую часть запроса (query) покрывает находка показано в колонке Query cover. Процент совпавших аминокислотных остатков - в Ident, а идентификатор и ссылка на запись в одной из баз данных – в Accession. E value – показатель возможности того, что найденное совпадение случайно. Для его определения анализируется банк случайно перемешанной исходной последовательности.
Участок совпадения – 212 аминокислот. Они находятся с 7 по 218 на исходной последовательности и с 3 по 214 на предполагаемом гомологе. При этом в выравнивании нет гепов. 165 аминокислот в выравнивании функционально похожи (77%, имеют положительный вес в матрице выравнивания), а 119 из них совпадают (56%).
Длина найденного белка - 214 аминокислот. Значит, 99% найденной последовательности похожи на запрос.
Это белок из той же системы Ktr, только белок С. Он также выделен из бактерии Bacillus subtilis.
Лучшая же находка представляет исходный белок. Соответственно последовательности полностью совпадают. Вес такой находки составляет 454, а E value – 5e-162, то есть почти 0.
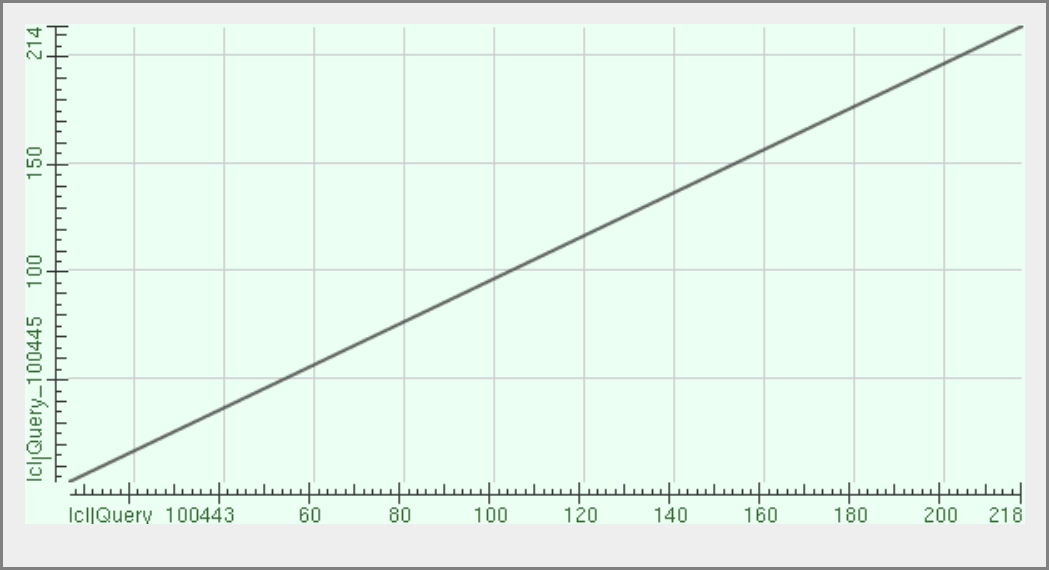
Алгоритм blastp позволяет также сравнивать последовательности, данные на вход. Например, строится карта локального сходства. Для исходной последовательности и выбранной находки такая карта представлена на рисунке 1.
Рисунок 1. Карта локального сходства между запросом и выбранной находкой (исходная последовательность расположена на горизонтальной оси).
Наличие единственного отрезка на такой карте коррелирует с тем фактом, что в выравнивании нет гепов. Соответственно, нет вставок и делеций. Этот длинный участок сходства и составляет участок гомологии последовательностей.
При поиске возможно указывать не только базу данных, в которой проводится поиск, но и таксон организмов. Для моего запроса нет никаких, сколько либо значительных, находок среди эукариот. Но есть среди архей. Характеристики такой находки представлены в таблице 2.
Таблица 2. Описание находоки blastp из архей
| Description | Max score | Total score | Query cover | E value | Ident | Accession |
|---|---|---|---|---|---|---|
| RecName: Full=Probable potassium channel protein 2; AltName: Full=MjK2 | 72 | 72 | 98% | 1e-13 | 26% | Q58752.1 |
Выравнивание этих последовательностей уже содержит гепы (11% выравнивания). Эти небольшие гепы отвечают вставкам и делециям, хорошо видным на локальной карте выравнивания (рисунок 2).
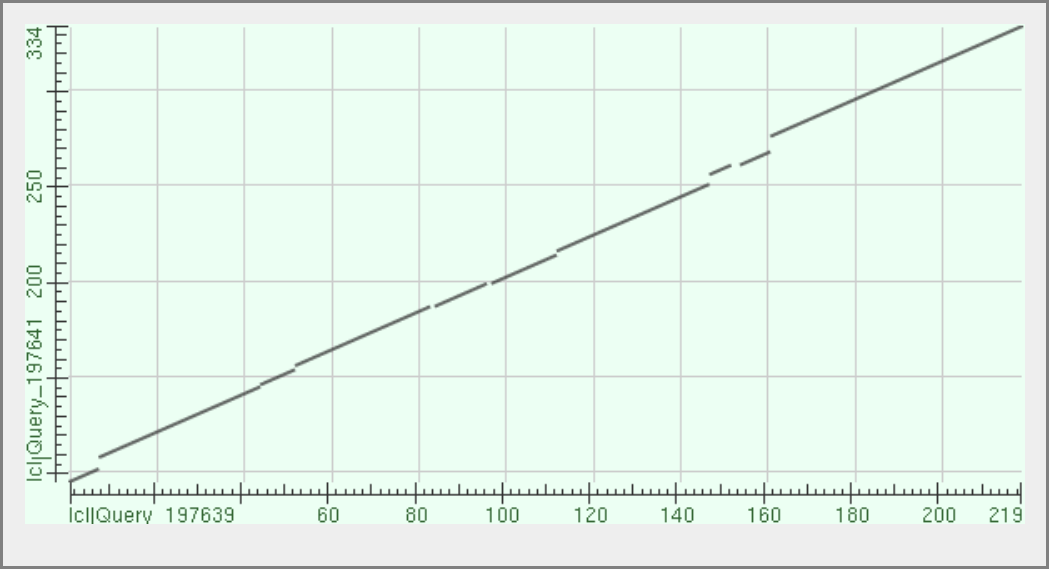
Рисунок 2. Карта локального сходства между запросом и находкой из архей (исходная последовательность расположена на горизонтальной оси).
Всего поиск по базе данных UniProtKB/Swiss-Prot дает 14 результатов, из которых только 3 (помимо исходной последовательности) имеют значимое для подтверждения гомологии E value (меньше 1e-3). Поэтому для построения множественного выравнивания я выполнил поиск по базе RefSeq_protein. При этом первые 100 находок имеют нормированный вес больше 200 и покрывают находку не менее чем на 95%.
Для множественного выравнивания я выбрал наиболее отличающиеся последовательности из разных групп бактерий. Из-за высокой степени гомологии выравнивание практически не содержит гепов. Кроме того, 59 колонок являются строго консервативными во всех 7 последовательностях при длине выравнивания 186 аминокислот, что составляет 30,1%. А функционально консервативны 119 колонок (60,7% выравнивания).
На рисунке 3 представлен фрагмент множественного выравнивания – первые 82 аминокислоты. Видно протяженный участок полной консервативности с 11 по 18 остатки.
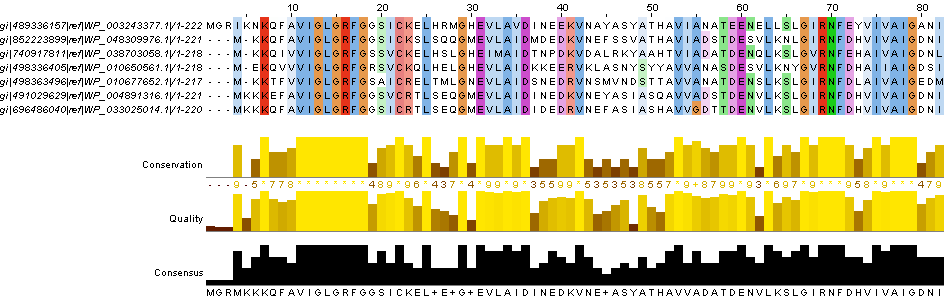
Рисунок 3. Фрагмент множественного выравнивания находок. Окраска Clustalx с консервативностью 50%.
Проект с выравниванием можно скачать тут.
Последнее обновление: 29.02.2016