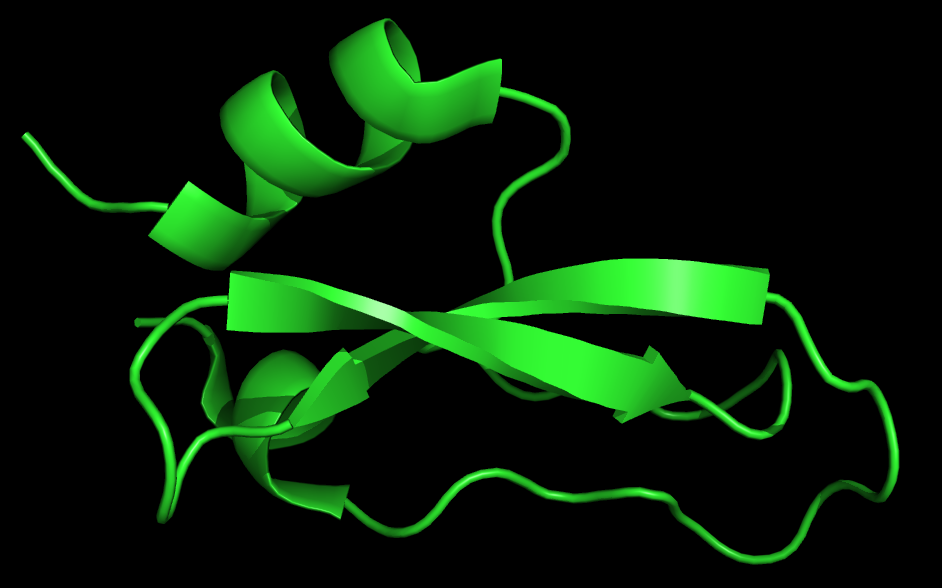

В качестве примера белка, структура которого была расшифрована обоими методами (РСА и ЯМР), был выбран бычий ингибитор трипсина. Длина этого белока всего 65 аминокислотных остатков. Структура белка полученная с помощью РСА имеет PDBid 1BPI (разрешение 1.09 Å), с помощью ЯМР - 1JV8. В 1JV8 записано 23 моделей. Структуры, визуализированные в PyMOL, представлены на рисунке 1.
|
|
Для сравнения двух моделей я выбрала 3 прдеполагаемые водородные связи в струкуктуре, полученной методом РСА (рисунок 2). Эти связи выбраны из разных структур белка: между атомами, отвечающими за стабильность альфа-спирали, (ARG_53/N - GLU_49/O, расстояние 3 Å), между боковыми группами на поверхности белковой глобулы (ASP_50/OD1 - ARG_53/NH2, расстояние 2.8 Å) и в ядре белка (ARG_1/NH2 - TYR_23/OH, расстояние 3.4 Å).
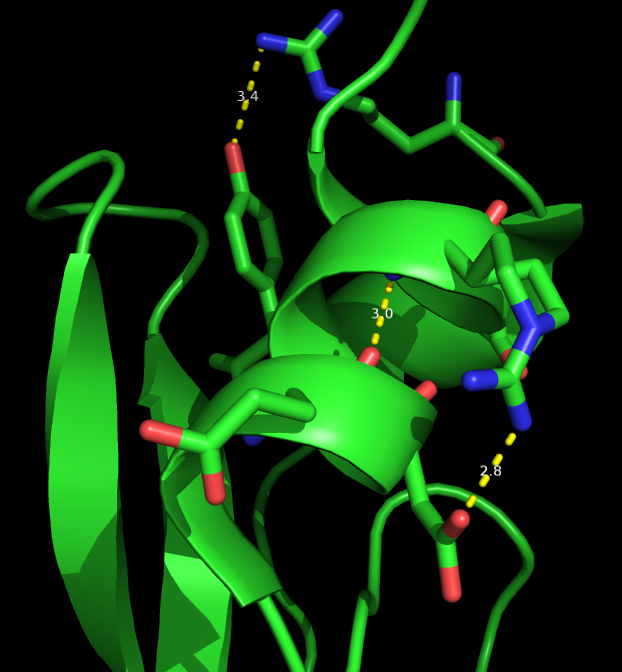
Критерием наличия водородной связи - расстояние между атомами меньше 3.5 ангстрем. Таблица расстояний для соотвествующих атомов 23 ЯМР моделей можно посмотреть здесь.
Водородные связи, удерживающие вторичную структуру должны быть наиболее устойчивыми. Это мы и видим на примере альфа-спирали. Расстояния (по 23 ЯМР моделям) между атомами (ARG_53/N - GLU_49/O) меняется в диапазоне: от 2.6 до 3.6 Å.
Медианы растояний между боковыми группами на поверхности белковой глобулы (ASP_50/OD1 - ARG_53/NH2) и вмежду атомами из ядра белка (ARG_1/NH2 - TYR_23/OH) соответственно 6.1 и 5.3 Å. А также можно видеть, что у них весьма большой разброс между минимальным значением и максимальным. Эти связи не стабилизированы окружающей их жесткой структурой, поэтому эти боковые группы могут занимать более вариабельные положения.
Последнее обновление: 10.12.2016