NGS практикум 11-14. Капшай Олег
Практикум 11
Для данного практикума мне был дан образец SRR10720404, который можно найти по ссылке https://www.ncbi.nlm.nih.gov/sra/SRR10720404. Секвенирование произыедено при помощи Illumina Genome Analyzer IIx, организм - Homo sapiens. Стратегия секвенирования - полно-экзомное секвенирование, чтения парноконцевые, ожидается 38,518,929 чтений.
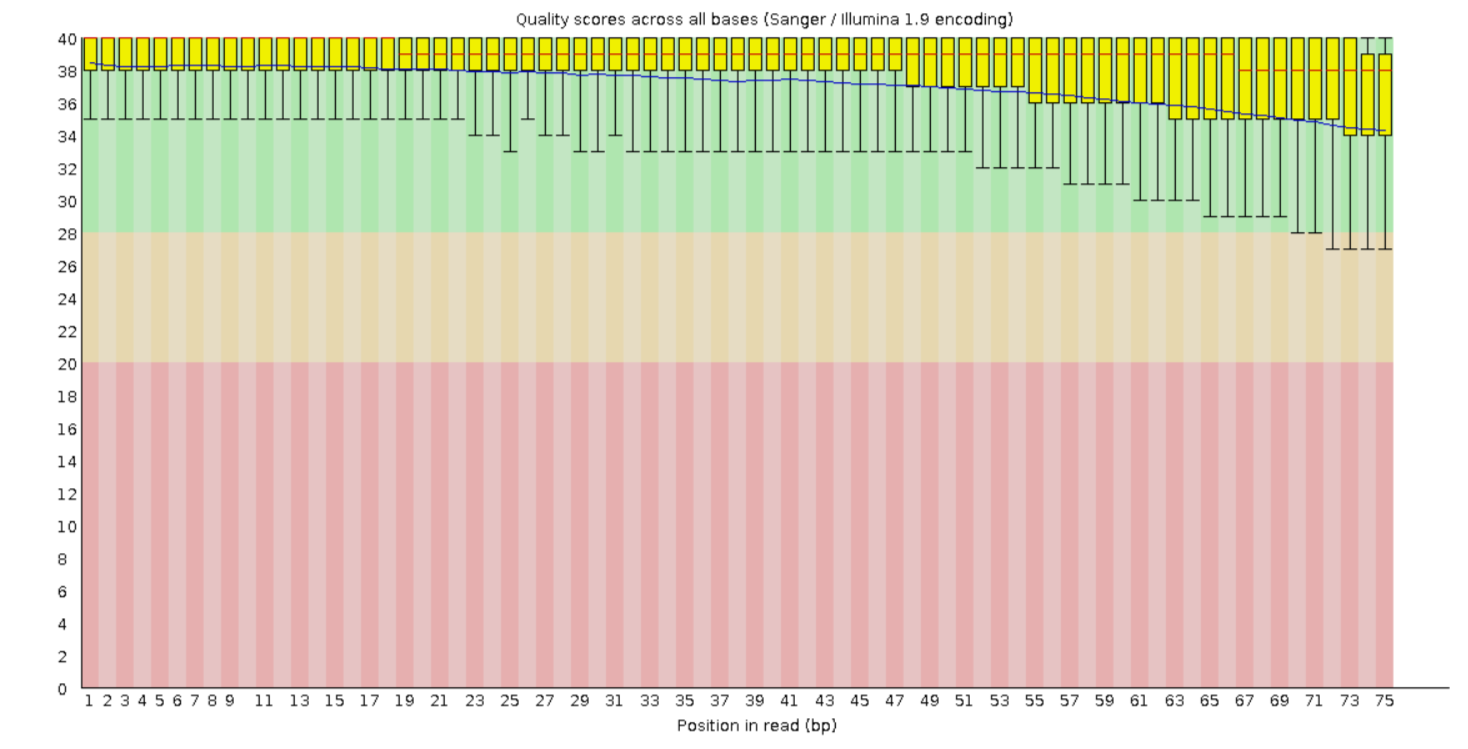
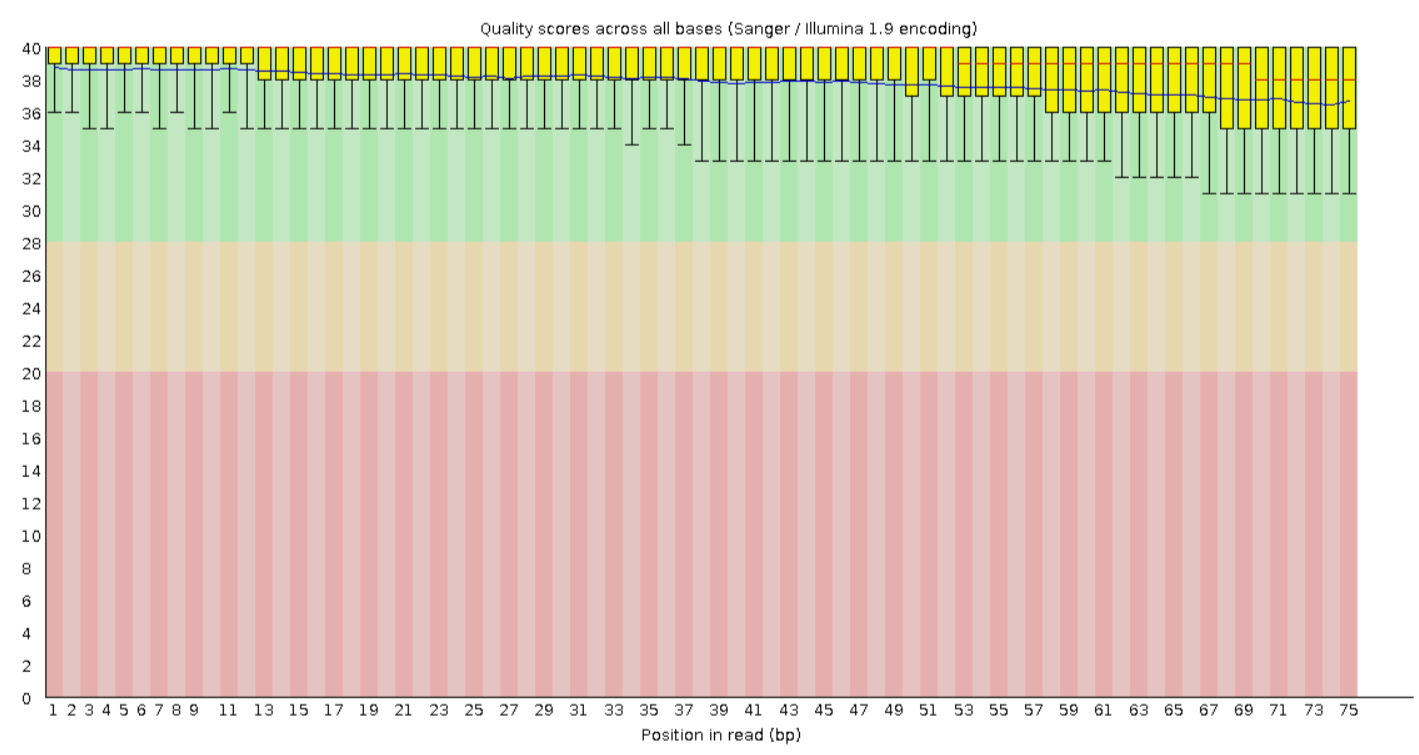
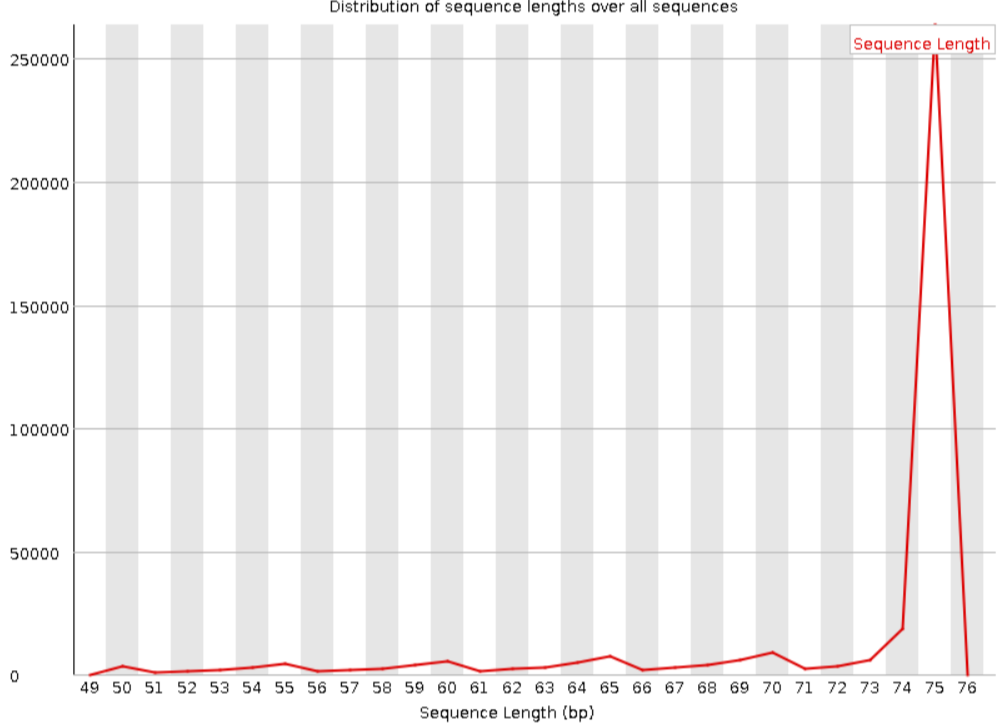
По результатам проверки программой fastQC, количество пар равно 38,518,929 (количество прямых и обратных чтений совпадает). На рисунках 1-2 представлено качество чтений по каждой позиции. Как видно из графиков, качество чтений довольно высокое. На рисунках 3-4 представлено распределение длин чтений, графики очень скучные, тк есть все чтения длиной 75 нуклеотидов.
Далее были отброшены нуклеотиды с конца с качеством 20 и меньше, оставлены варианты не короче 50 нуклеотидов при помощи команды:
$TrimmomaticPE SRR10720404_1.fastq.gz SRR10720404_2.fastq.gz output_forward_pair ed.fq.gz output_forward_unpaired.fq.gz output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz TRAILING:20 MINLEN:50.
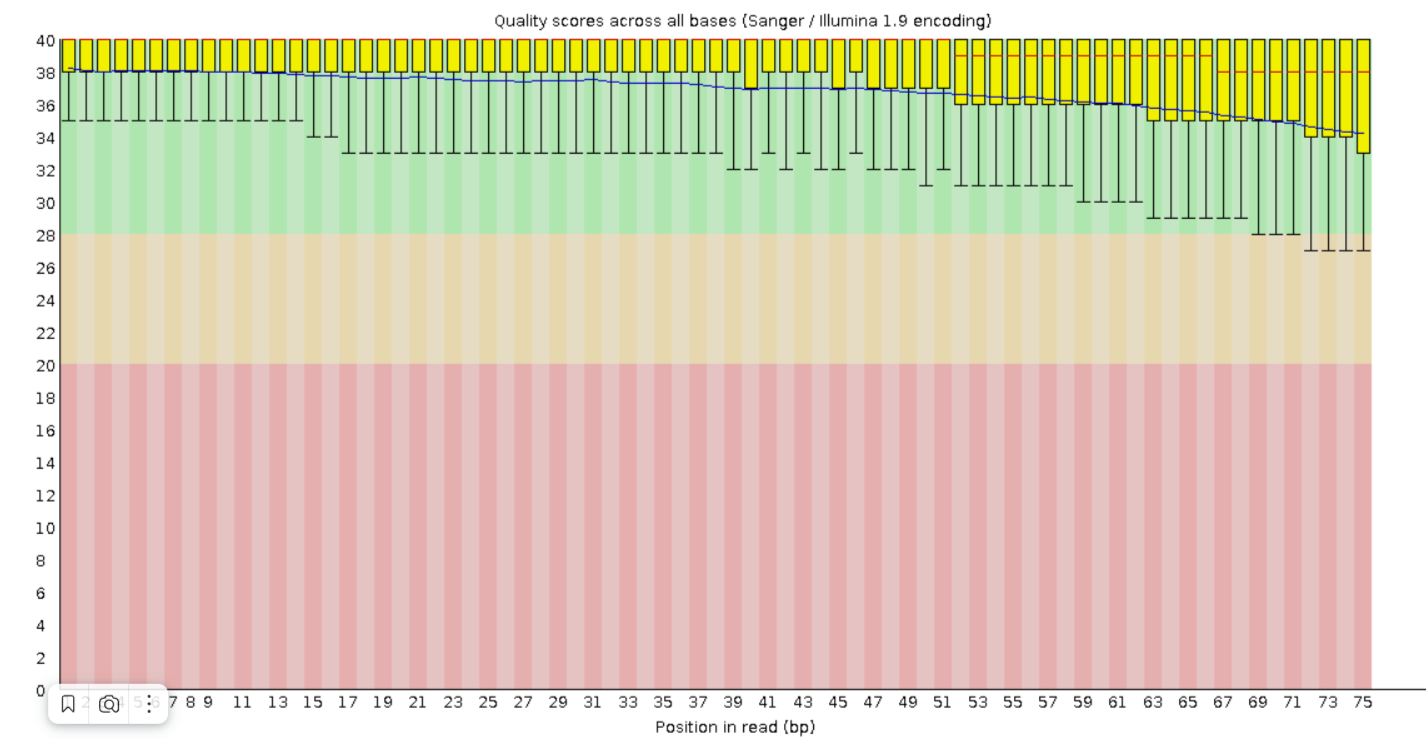
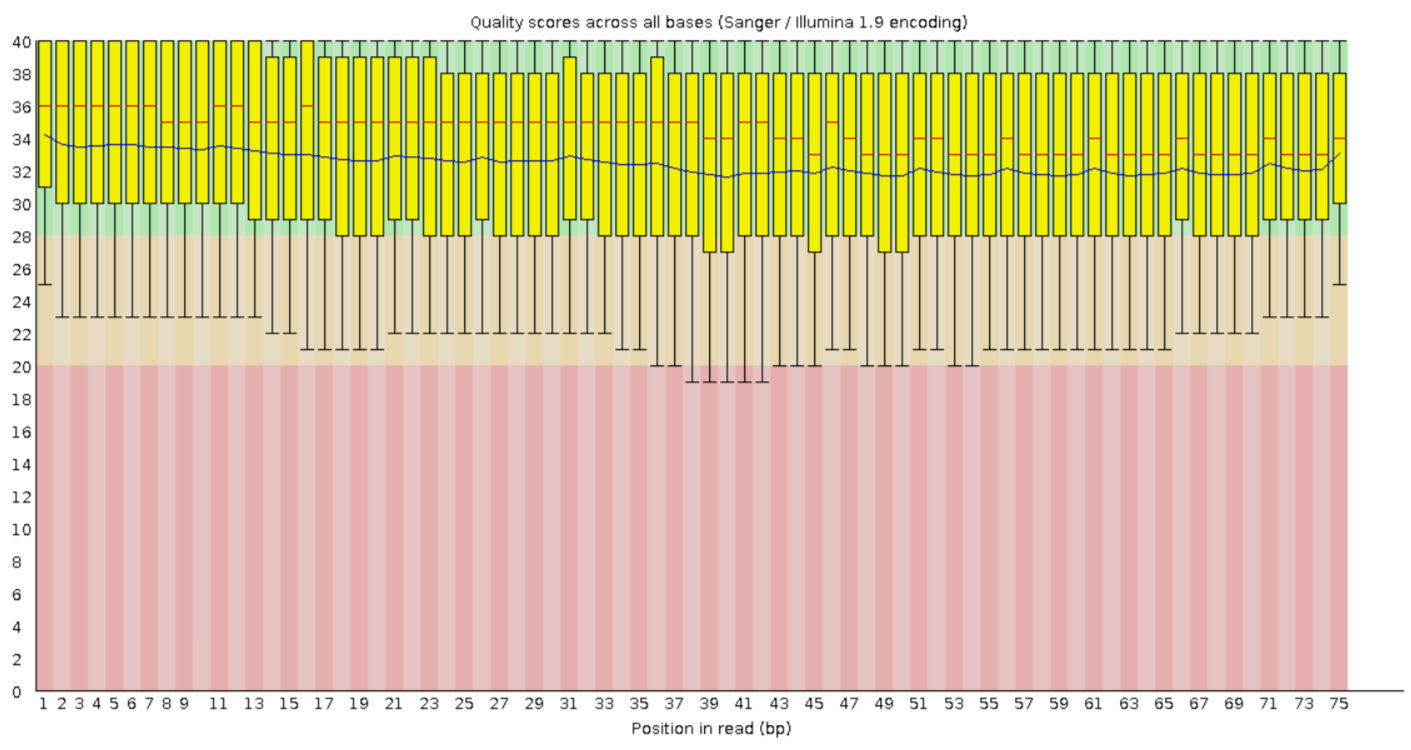
После обработки осталось 37,136,668 пар (96,41 %). Как можно видеть на рисунках 5-6, качество оставшихся пар (paired) увеличилось, теперь весь boxplot лежит в зеленой области. А в группу unpaired, наоборот, попали чтения с низким качеством. После тримирования поменялось распределение длин чтений, что видно на рисунке 7.
Практикум 12
В дальнейшем картирования были картированы на референсный геном. Все команды (вообще все) находятся на сервере kodomo, файл /mnt/scratch/NGS/kaps/assembly/code. После картирования при помощи hisat2 получился SAM файл весом 15 Гб, который был конвертирован в BAM файл весом 4,0 Гб. Всего на референс рактировалось 4,420,795 ридов (5.95 % от тримированных), из которых 3,871,328 корретно спарились (5.21 %). Далее были отобраны чтения, картированные на 5 хромосому в правильных парах. В полученном файле в корректных парах картировалось 3,871,328 чтений (100 %).
Практикум 13
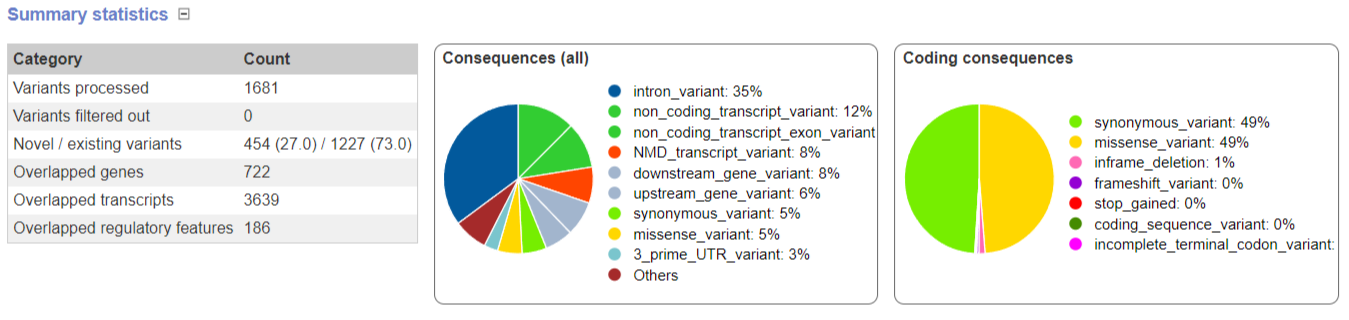
Все команды по прежднему хранятся в файле на кодомо. в практикуме 13 из полученных картировок искались варианты. При помощи bcftools было выявлено 84,733 варианта, из которых 82,231 - однонуклеотидные замены, 2,502 - вставки и делеции. Далее варианты были отфильтрованы, после чего осталось только 1,681 замена (2.04 %), их них 1,639 однонуклеотидных замен (1.99 %) и 42 вставки/делеции (1.68 %). На рисунке 8 представлена аннотация вариантов при помощи VEP. К сожалению, ни один вариант не получил IMPACT HIGH. Довольно интересно, что количество синонимичных и миссенс мутаций примерно равно.
Практикум 14
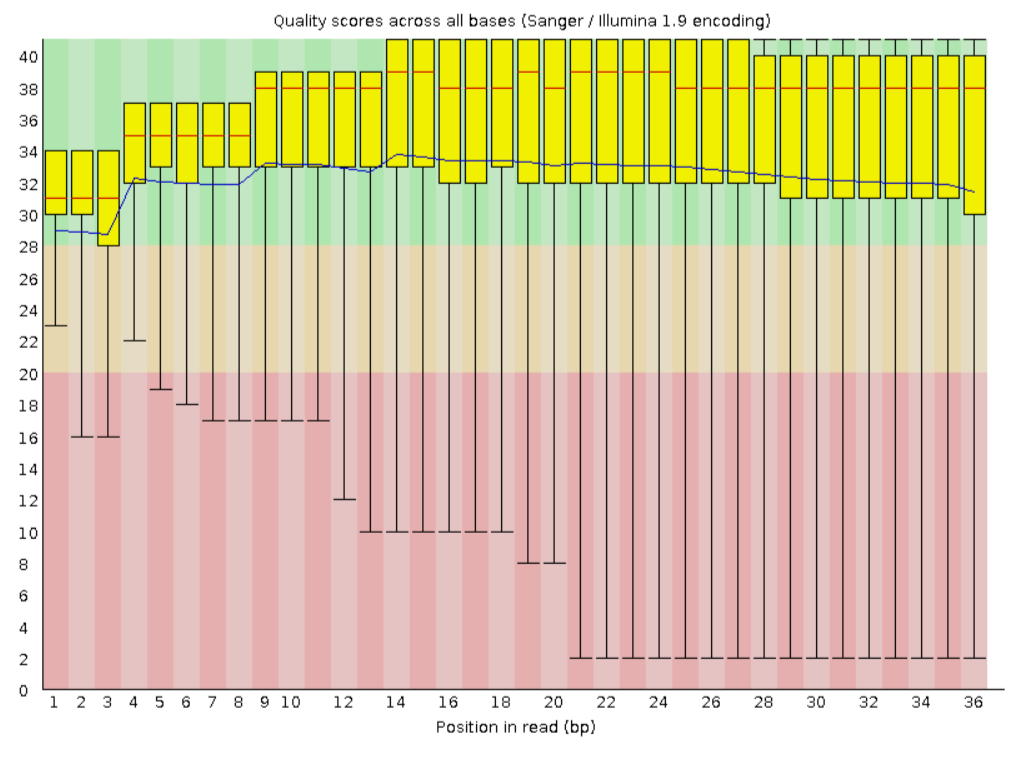
Для 14 практикума мне был дан образец с ID ENCFF038OLY, ссылка на за пись в базе данных https://www.encodeproject.org/experiments/ENCSR096USV/. Образец взят из мышечной ткани ноги 127 дневного эмбриона человека. Стратегия секвенирования - тотальная РНК, чтения одноконцевые, цепь-специфичности нет. Качество чтений было проверено при помощи fastQC, результаты на рисунках 9-10. Всего 72,517,664 чтения. Как видно из рисунка, большиство чтений имеют высокое качество, но разброс большой. Также видно, что боксплот у программы имеет довольно странные усы, которые вообще не похожи на полтора межквартильных расстояния (как у нормального боксплота). Рисунок 10 не дает ни какой интересной информации, все чтения имеют длину 36 (мы могли это увидеть в разделе с базовой статистикой).
При помощи hisat на 5 хромосому картировалось 5,488,466 чтений. Команда для картирования:
$hisat2 -x genes.gtf -k 3 -U ENCFF038OLY.fastq.gz
Далее был произведен поиск экспресирующихся генов. Для этого был использован файл разметки генома, который представляет собой tsv таблицу, которая содержит название участка, его координаты, цепь и другие парметры. Для каждого гена считается количество картировок при помощи htseq-count. Параметр -t отвечает за тип последовательности, которую мы считаем в геноме (в моем примере экзоны). -f - формат файла, -s - являются ли данные цепь-специфичными. -m - что делать, если чтение легло на более чем один участок (в нашем случае union). Итоговая команда:
$htseq-count -t exon -f bam -s no -m union rna.5.bam ../reference/genes.gtf
В результате 1,064,576 чтений попало в границы генов. Соответственно мимо границ генов попало 4,423,890 чтений. Это можно объяснить наличием существованием огрномного количества некодирующих РНК.