На страницу четвертого семестра
Моделирование эволюции гена
Графическое изображение дерева
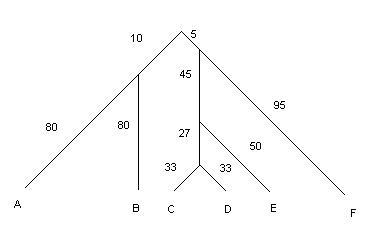
Видно, что данное дерево не является ультраметрическим,
так как не все расстояния от
корня до листьев одинаково.
Скобочная форма:
((А:80,В:80):10,(((С:33,D:33):27,Е:50):45,F:95):5)
;
Длина гена: 1518 нуклеотидных остатка.
Топология дерева
Топология дерева может быть изображена в форме таблицы:
| A
|
B
|
C
|
D
|
E
|
F
|
| .
|
.
|
*
|
*
|
*
|
*
|
| .
|
.
|
*
|
*
|
*
|
.
|
| .
|
.
|
*
|
*
|
.
|
.
|
Построение эволюционной модели
Цель - получить последовательности, стоящиев узлах и листьях дерева, приведенного выше;
при условии что в позиции (1) стоит ген белка SYK1_Ecoli.
Скрипт, позволяющий получить мутантные последовательности:
msbar syk1_ecoli_gene1.fasta ab.fasta -point 4 -count 153 -auto
msbar ab.fasta A.fasta -point 4 -count 1221 -auto
msbar ab.fasta B.fasta -point 4 -count 1221 -auto
msbar syk1_ecoli_gene1.fasta cdef.fasta -point 4 -count 75 -auto
msbar cdef.fasta cde.fasta -point 4 -count 687 -auto
msbar cde.fasta cd.fasta -point 4 -count 412 -auto
msbar cd.fasta C.fasta -point 4 -count 504 -auto
msbar cd.fasta D.fasta -point 4 -count 504 -auto
msbar cde.fasta E.fasta -point 4 -count 764 -auto
msbar cdef.fasta F.fasta -point 4 -count 1451 -auto
Реконструкция дерева алгоритмами UPGMA, Neighbor-joining и Maximum Likelihood (максимального правдоподобия).
Цель задания – построить деревья на основе данных конечных
последовательностей (листьев), и сравнить результаты.
В результате реконструкции дерева алгоритмом максимального правдоподобия с помощью программы fdnaml
(команда fdnaml all.fasta -ttratio 1 -auto) было получено следующее
"текстово-графическое" изображение дерева:
+--------------------b
|
| +---------------------f
1---4
| | +----------e
| +-------------3
| | +-------d
| +-------2
| +-------c
|
+----------------a
Чтобы реконструировать дерево алгоритмами UPGMA или Neighbor-joining, сначала надо посчитать попарные расстояния между
последовательностями программой fdnadist:
fdnadist all.fasta -ttratio 1 -auto.
После этого этот файл был подан на вход программе fneighbor:
(fneighbor all.fdnadist -auto
для реконструкции алгоритмом Neighbor-joining и
fneighbor all.fdnadist -treetype u -auto
для реконструкции алгоритмом UPGMA).
В результате получили деревья:
Для Neighbor-joining:
+--------------------b
!
! +--------c
! +--------1
! +--------------2 +-------d
! ! !
3---4 +----------e
! !
! +----------------------f
!
+----------------a
Для UPGMA:
+------------------------------------a
+--------3
+-----4 +------------------------------------b
! !
! +---------------------------------------------f
--5
! +---------------c
! +----------1
+------------------------2 +---------------d
!
+--------------------------e
Сравнение реконструированных деревьев между собой и с правильным деревом
Для сравнение предложено было сделать таблицу, в левой части которой приведены (в виде точек и звёздочек) все ветви,
встреченные во всех деревьях (исходном и трёх реконструкциях), а в правой добавлены четыре столбца, соостветствующие
четырём деревьям. Знаком + отмечено, в каких деревьях встретилась каждая из ветвей.
| A | B | C | D | E | F |
правильное дерево | 1ое | 2ое | 3ее |
| . | . | * | * | * | * |
+ | + | + | + |
| . | . | * | * | * | . |
+ | + | + | + |
| . | . | * | * | . | . |
+ | + | + | + |
Несмотря на то, что в данном случае все алгоритмы выдали правильный результат, надежнее было бы использовать
методы UPGMA и Neighbor-joining
Бутстреп-анализ выравнивания мутантных последовательностей
Цель задания – получить консенсусное дерево с помощью методa
bootstrep и сравнить его с реальным деревом.
Деревья получаются путем последовательного применения трех программ из пакета PHYLIP:
- seqboot
- dnaml
- consense
Выходной файл all.fconsense содержит следующее неукорененное дерево:
+--------------------e
|
+100.0-| +------b
| | +-93.0-|
| +100.0-| +------a
+------| |
| | +-------------f
| |
| +---------------------------d
|
+----------------------------------c
Реальные данные:
Результаты бутстеп-анализа:
| A | B | C | D | E | F | сколько раз встречается (из 100) |
| . | . | * | * | * | * | 93 |
| . | . | * | * | * | . | 100 |
| . | . | . | . | * | * | 100 |
Из полученных данных видно, что все ветви имеют высокую степень надежности.
Топология воссоздана правильно.
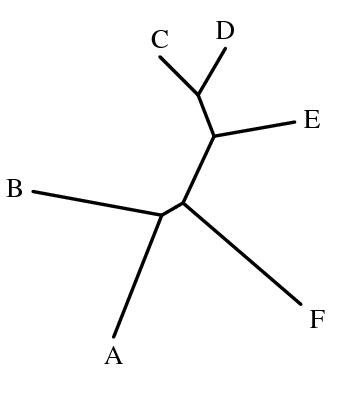
Создание изображения своего дерева программой fdrawtree
Скобочная формула была помещена в отдельный файл, который был подан на вход программе fdrawtree. В
результате было получено следующее изображение:
©
Долудин Юрий, 2005