Практикум №15
Сборка de novo.
1. Подготовка чтений с помощью программы Trimmomatic.
Код доступа проекта по секвенированию бактерии Buchnera aphidicola, который был предоставлен мне, - SRR4240378. Перейдя на сайт ENA, я скачать соответствующий файл в формате fastq с использованием следующей команды:
wget ''ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/008/SRR4240378/SRR4240378.fastq.gz''
Далее я решил создать файл в формате fasta, содержащий все возможные последовательности адаптеров, которые необходимо вырезать из предоставленных прочтений. Это было сделано следующей командой:
cat /mnt/scratch/NGS/adapters/* > adapters.fasta
Следующей командой я удалил перечисленные в файле adapters.fasta адаптеры из чтений в файле SRR4240378.fastq.gz посредством программы Trimmomatic:
java -jar /usr/share/java/trimmomatic.jar SE -threads 10 -phred33
-trimlog trim.log SRR4240378.fastq.gz SRR4240378_trimmed_adapt.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7
После работы программы количество чтений сократилось с 4420587 до 4338744. То есть 81843 (1.85%) чтений было удалено.
Далее я удалил с 3'-конца чтений нуклеотиды, которые не прошли порог качества (то есть имели качество меньше 20), сохраняя при этом минимальную длину в 32 нуклеотидов:
java -jar /usr/share/java/trimmomatic.jar SE -threads 10 -phred33 -trimlog trim2.log SRR4240378_trimmed_adapt.fastq.gz SRR4240378_full_trimmed.fastq.gz TRAILING:20 MINLEN:32
При этом теперь из 4338744 чтений осталось 4154738. Было отброшено 184006 чтений (4.24%)
Исходный файл (SRR4240378.fastq.gz) имел размер в 91M, после обрезки адаптеров размер сократился до 89M (SRR4240378_trimmed_adapt.fastq.gz), а после обрезания 3'-конца упал до 84M (SRR4240378_full_trimmed.fastq.gz)
2. Формирование k-меров с помощью velveth.
Затем я сформировал директорию (Assembly_SRR4240378) с k-мерами (k-мерами длиной 31 в нашем случае) из обработанных чтений с помощью следующей команды:
velveth Assembly_SRR4240378 31 -short -fastq.gz SRR4240378_full_trimmed.fastq.gz
3. Сборка посредством velvetg.
И, наконец, сама сборка была осуществлена командой velvetg, требующей в стандартном случае на вход лишь директорию, содержащую k-меры, подготовленные на предыдущем шаге:
velvetg Assembly_SRR4240378
Показатель N50 принял значение 7028.
Длины трёх самых длинных контигов - 36746, 19371 и 16745. Им соответствуют покрытия 20.02, 20.54 и 20.90.
С помощью сортировки по столбцу с показателем coverage (из файла stats.txt) я нашёл последовательности с наименьшим и наибольшим coverage, длина которых больше 2k (62 bp), поскольку только такие последовательности отображаются в файле contigs.fa. Ниже приведены их заголовки из файла contigs.fa:
NODE_166_length_64_cov_2.843750
NODE_81_length_934_cov_102.748390
4. Анализ контигов.
На рисунке №1 представлен DotPlot, полученный в результате выравнивания самого длинного контига (36746) на геном Buchnera aphidicola с AC CP009253 с помощью алгоритма megablast. Контиг картировался по семи участкам, характеристики выравниваний которых я привожу ниже.
Фрагмент генома
Identities
Gaps
480874 - 481545
564/686(82%)
20/686(2,9%)
481997 - 488106
4621/6238(74%)
308/6238(4,9%)
493487 - 494864
1109/1384(80%)
13/1384(0,94%)
495033 - 495148
108/120(90%)
5/120(4,2%)
496111 - 500325
3255/4324(75%)
154/4324(3,6%)
500370 - 508806
6516/8617(76%)
351/8617(4,1%)
510438 - 516539
4897/6234(79%)
187/6234(2,9%)
Фрагмент генома
Identities
Gaps
573092 - 582686
7212/9822(73%)
461/9822(4%)
584329 - 587055
2100/2777(76%)
108/2777(3%)
Фрагмент генома
Identities
Gaps
144368 - 151796
5863/7536(78%)
243/7536(3%)
Как видно, этот контиг лучше всего картируется на геном (единым участком), хоть и не имеет никакого преимущества в идентичности (количество совпадающих оснований).
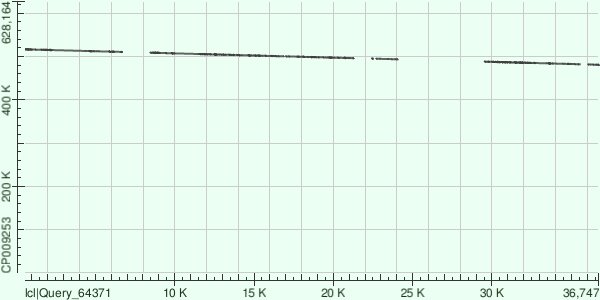
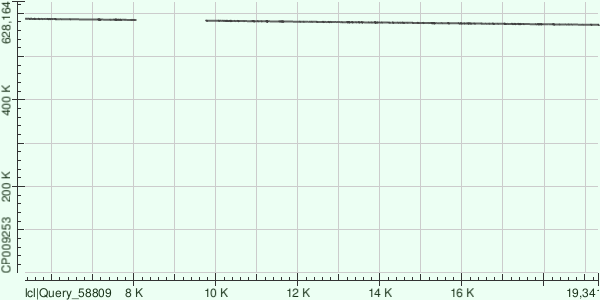
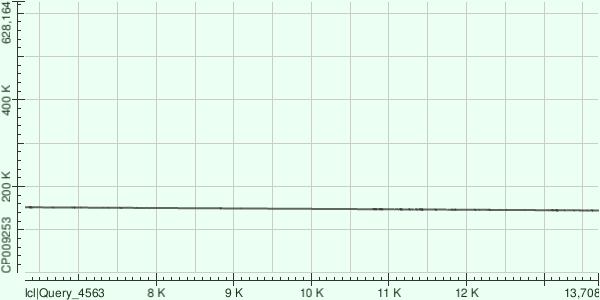
В целом можно сказать, что самый большой контиг картировался на геном наименее идеально, сформировав 7 отдельных выравниваний, одно из которых имеет протяжённость всего лишь в 120 нуклеотидов.
Кирилл Кузенков, студент второго курса ФББ