Практикум №4: AlphaFold
Задание A8: Амилоиды.
В ходе выполнения первой задачи в центр внимания ставится вопрос о качестве предсказания нашумевшей нейросетьевой моделью AlphaFold ненативных белковых структур, называемых амилоидами. Амилоид - это скопление неправильно сложенных белков, принимающих форму так называемой амилоидной фибриллы, в которой β-листы, расположенные перпендикулярно оси фибриллы, складываются в стопку и, взаимодействуя друг с другом, стабилизируют образованную структуру.
С помощью фреймворка BLAST удобно было идентифицировать происхождение выданного участка аминокислотной последовательности (A8: LYVGSKTKEGVVHGVATVAEKTKEQVTNVGGAVVTGVTAVAQKTVEGAGSIAAATGFV). Со 100% Identity было установлено сходство с белком α-синуклеином, локализованном преимущественно в ядрах нейронов головного мозга млекопитающих [1]. Данные свидетельствуют о том, что α-синуклеин - участник процесса синаптической активации, действующий в качестве молекулярного шаперона при образовании комплекса SNARE [2].
В случае гиперэкспрессии или наличия некоторых мутантных форм α-синуклеин способен формировать нерастворимые тельца включения, приводя к нейродегенеративным заболеваниям, названным синуклеинопатиями [3]. Наиболее известная из них - болезнь Паркинсона.
Являясь белком, состоящим из 140 АО (Рис. 1), α-синуклеин в том числе содержит центральную гидрофобную область (с 61 по 95 остатки), включающую область неамилоидного β-компонента, участвующую в агрегации белков [4].

В банке PDB была найдена структура мономера α-синуклеина (1XQ8), полученного с помощью ЯМР. Выданный мне участок представляет из себя фрагмент 38-95 (Рис. 1), включающий в себя центральную гидрофобную область, вовлечённую в формирование амилоидных фибрилл.
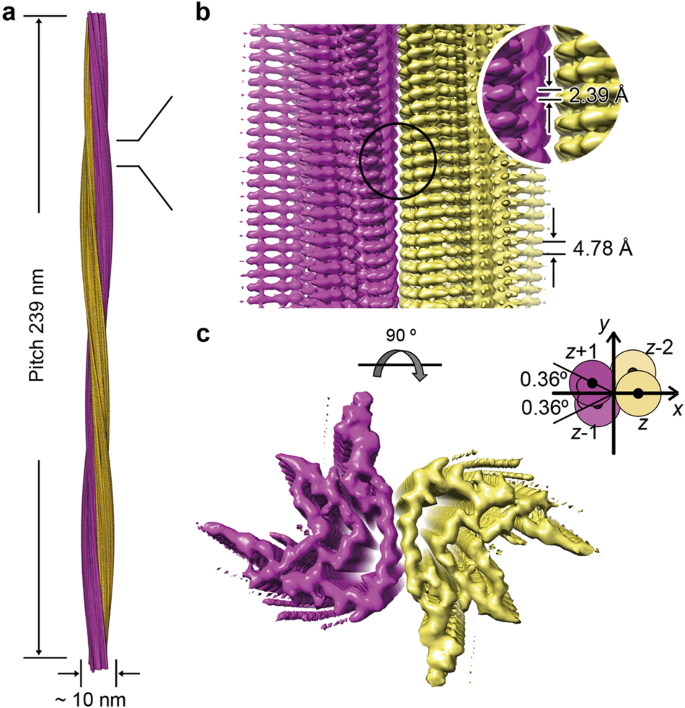
Также была найдена структура (получена методом КриоЭМ с разрешением 3.07 Å) самой амилоидной фибриллы (6A6B), гомо 12-мера, в каждом мономере которой отображены остатки 37-99, принимающие ключевое участие в её формировании (Рис. 2).
Амилоидная фибрилла α-синуклеина в качестве структурной единицы демонстрирует два протофиламента, переплетающихся вдоль 21-винтовой оси (поворот на π со сдвигом на полпериода). Каждый протофиламент (из 6 мономеров) напоминает организацией греческий ключ (Рис. 2).
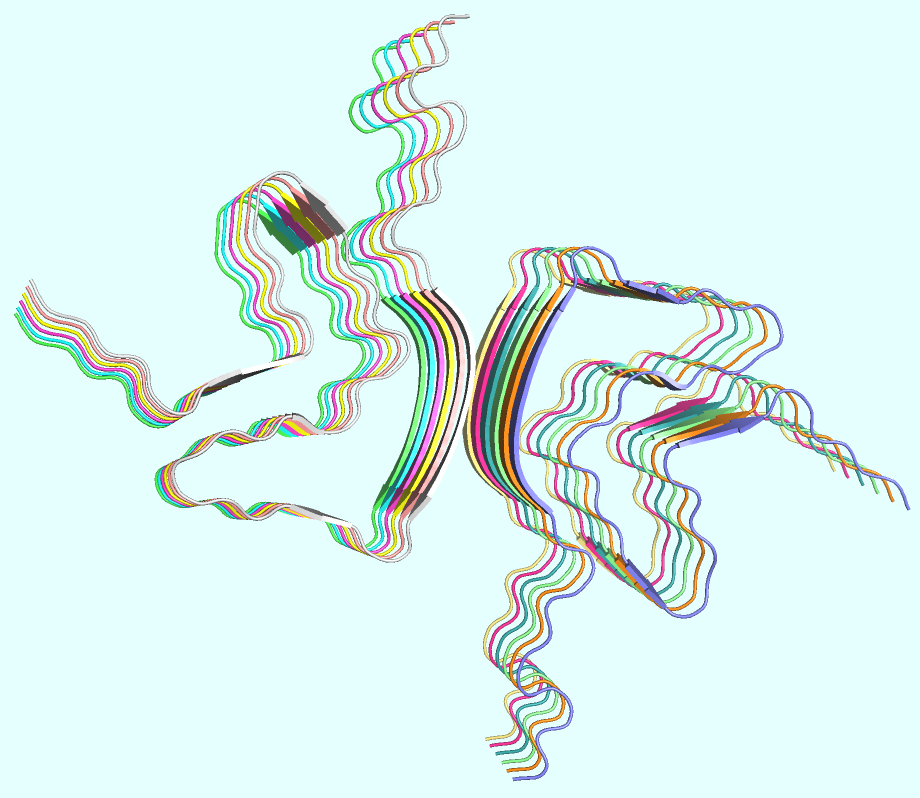
Полагаю, что AlphaFold вряд ли сможет хорошо предсказать устройство такого интересного протофиламента =) А вот у мономера есть все шансы.
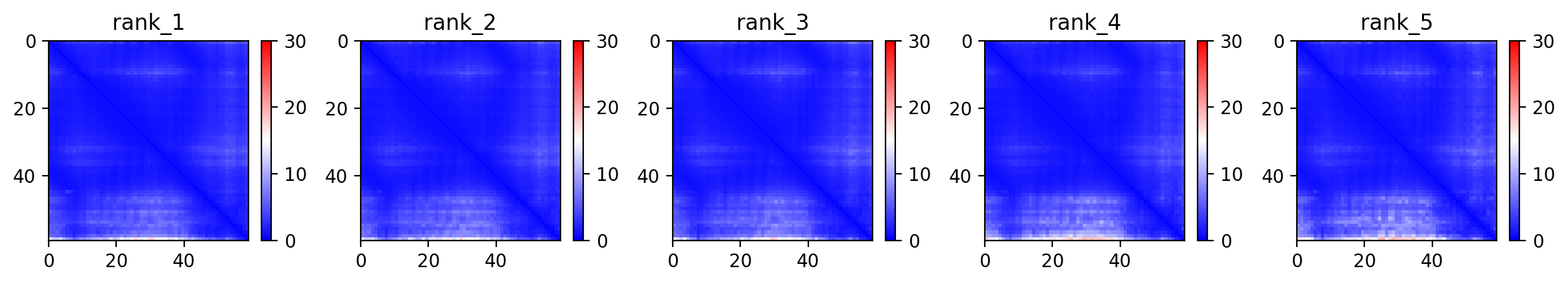
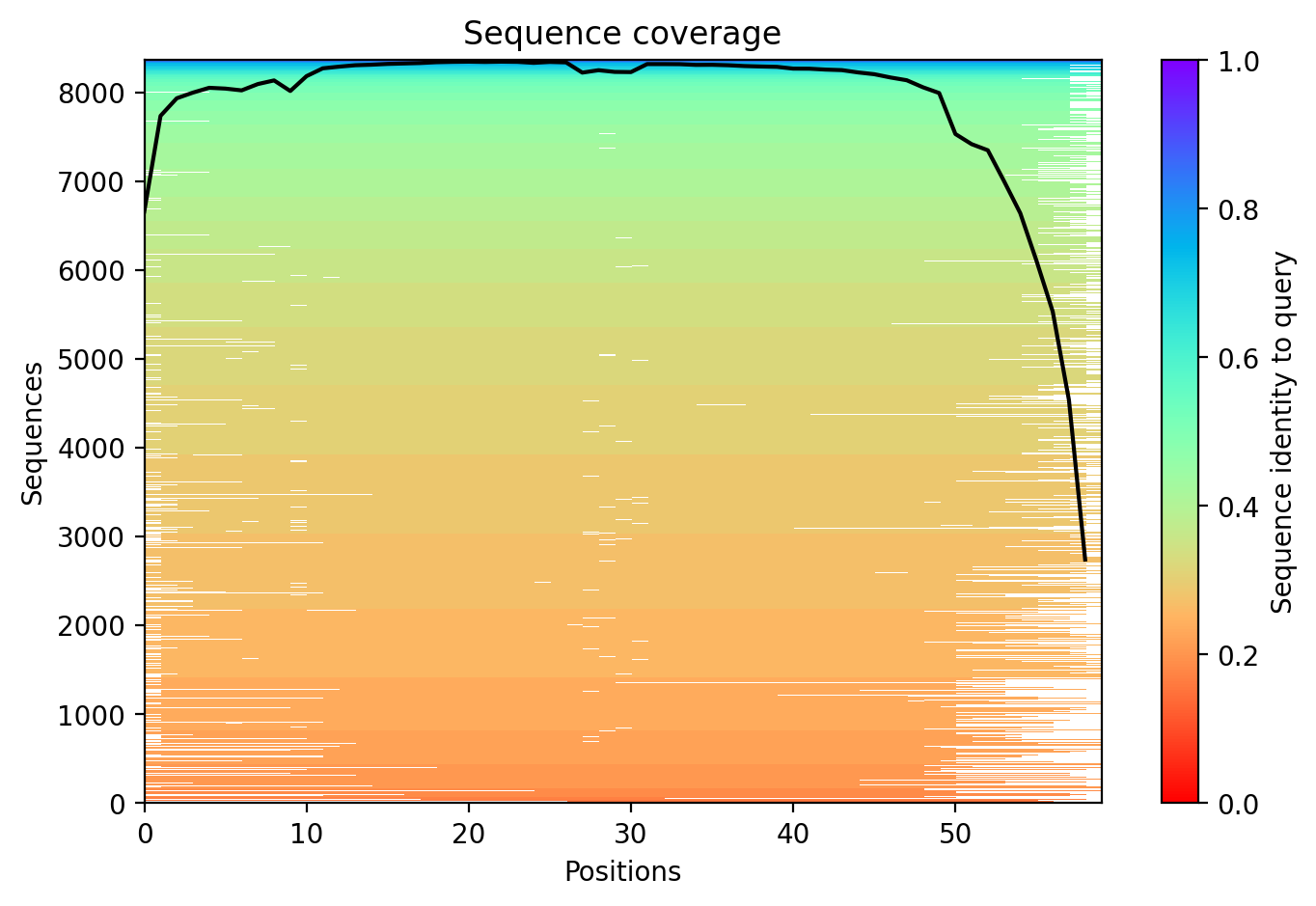
Посмотрим на метрики предсказания мономера (Рис. 3): PAE (Predicted Aligned Error - ожидаемая ошибка позиционирования для каждого из остатков в каждом "блоке". В данном случае блок один - мономер. Для пентамера будет 5 - A, B, C, D, E; для декамера - 10), pIDDT - знаменитая степень уверенности AlphaFold в своём предсказании (приводится для каждой из 5 построенных моделей: rank 1-5) и sequence coverage - соответствие последовательности запросу (для каждого блока).
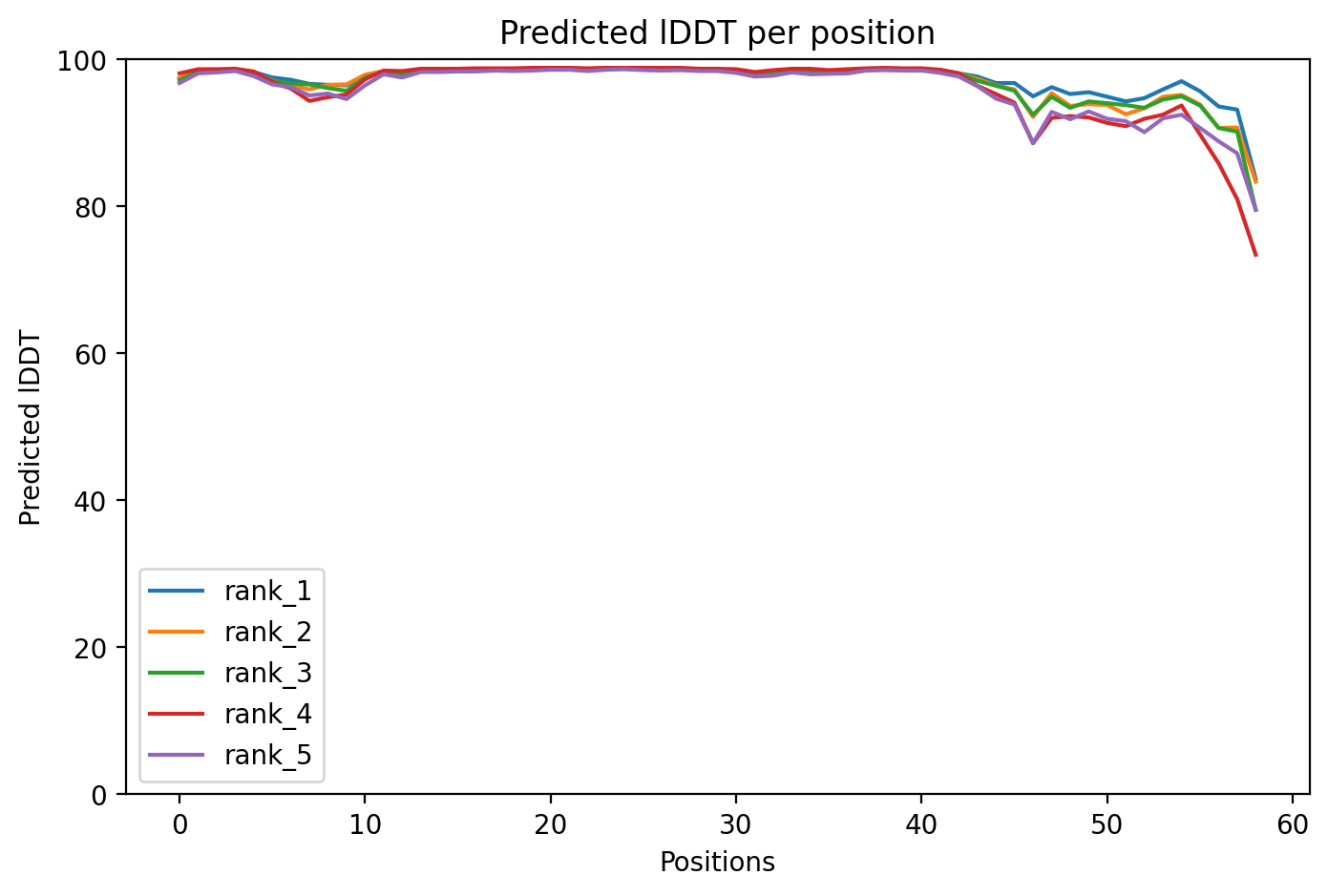
Как видим, метрики говорят о хорошем... Давайте рассмотрим структурное выравнивание модели (они все примерно одинаковые, но мы возьмём rank 1) со структурой 1XQ8. При этом вряд ли стоит ожидать точного сходства, ведь 1XQ8 - это весь белок (140 АО), а структура модели - это лишь участок белка (38-95), который сам по себе будет сворачиваться, конечно, иначе в отсутствие контекста всего белка (Рис. 4).
И, действительно, результат получается странным: модель содержит β-листы, которые в экспериментальной структуре отсутствуют. Это понятно, поскольку выбранный фрагмент содержит центральную гидрофобную область белка, который имеет все потенции для образования таких β-листов. Но в большом количестве эти β-листы образовываются только при взаимодействии большого количества мономеров. В отдельном же мономере их образование оказывается энергетически невыгодным.

Рассмотрим предсказание пентамера. Тургенев сказал бы:"Мимо, читатель, мимо!"... Как и предполагалось, предсказания вышли "с особенностями". Степень уверенности модели (pIDDT), PAE и Coverage (Рис. 5) оказались неудовлетворительными. pIDDT
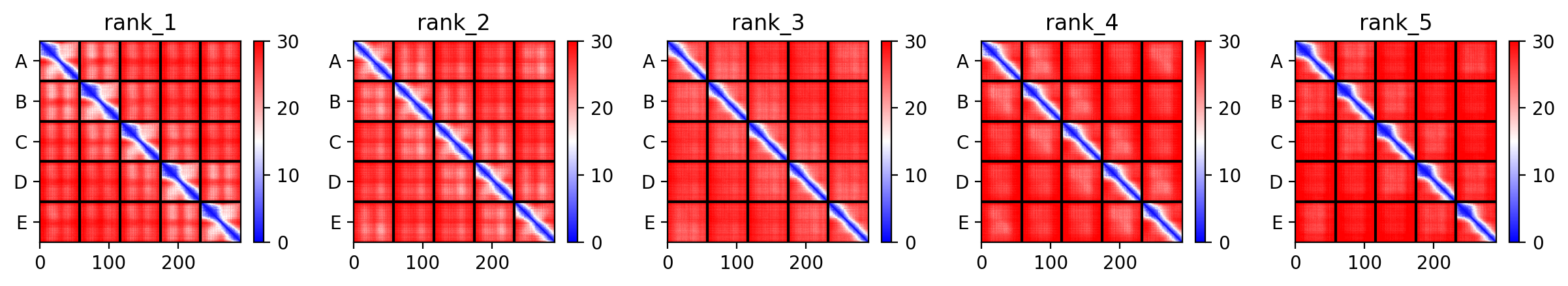
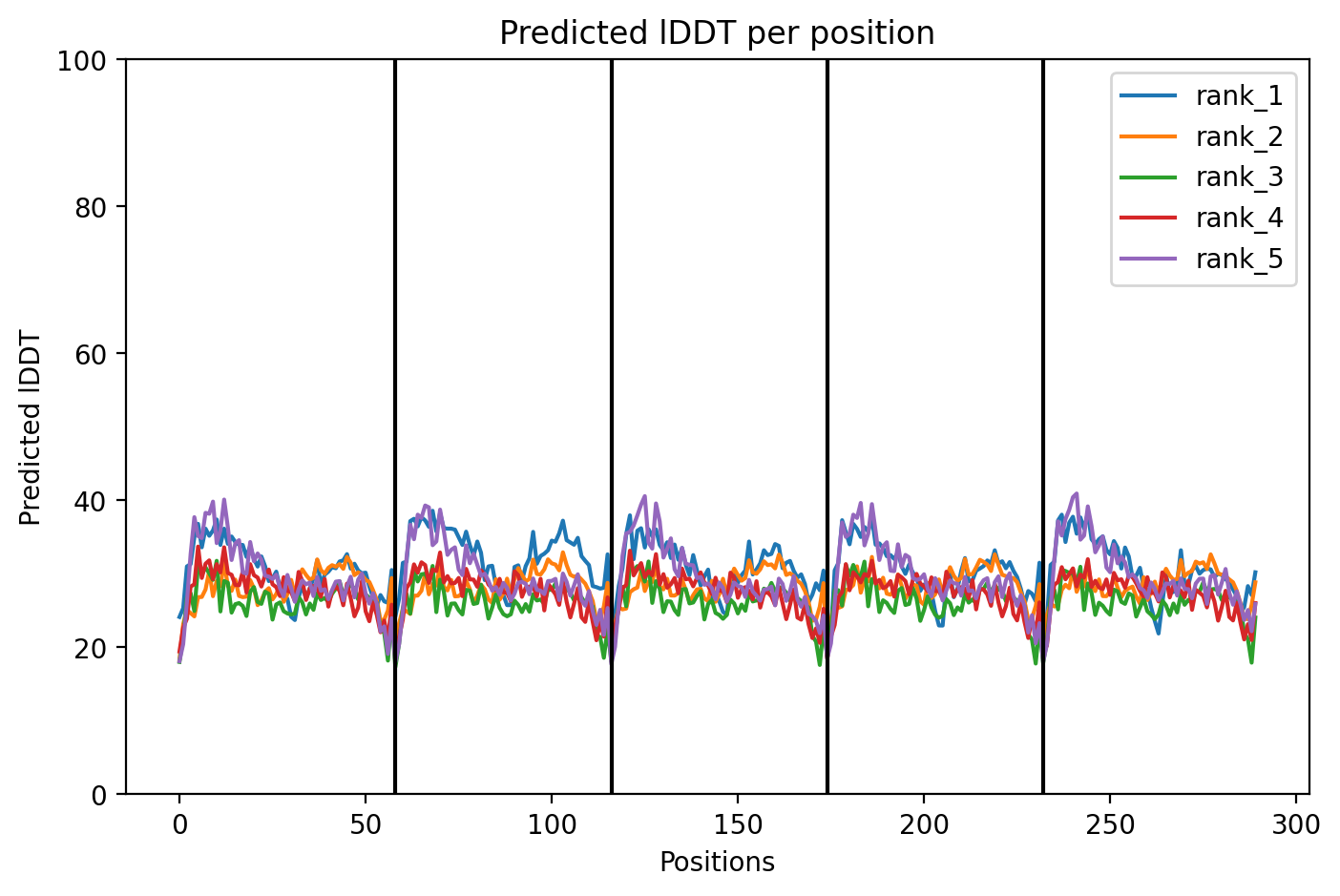
Аналогичная картина разворачивается с метриками при предсказании декамера (Рис. 6).
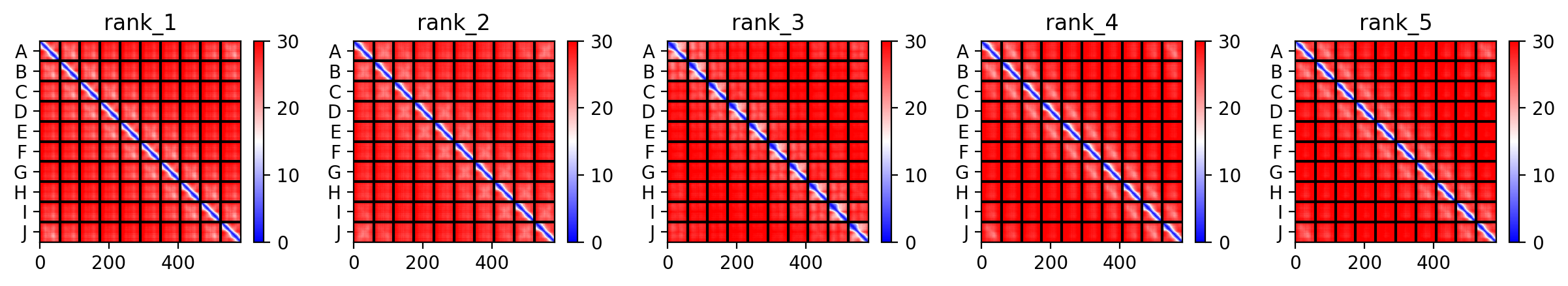
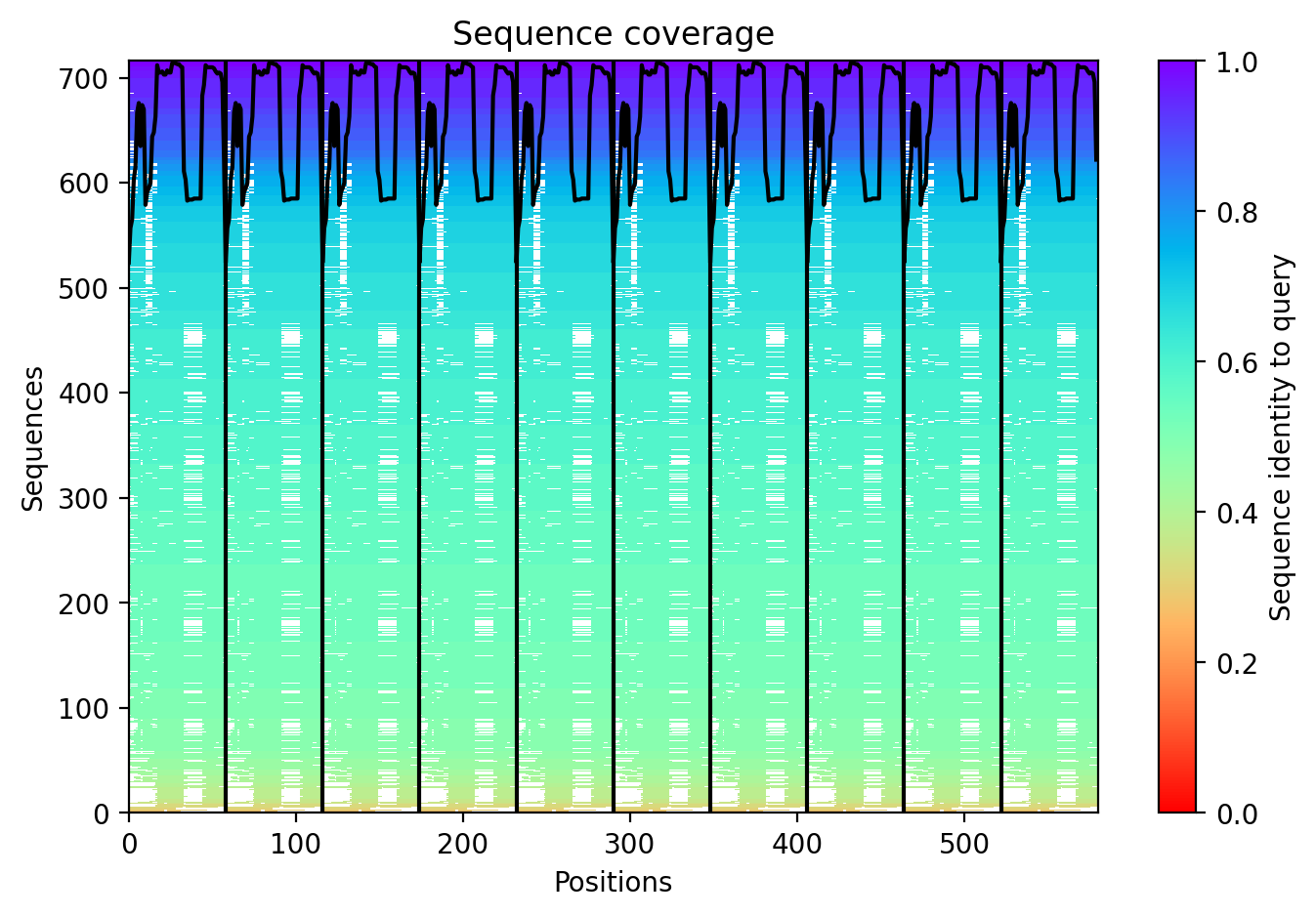
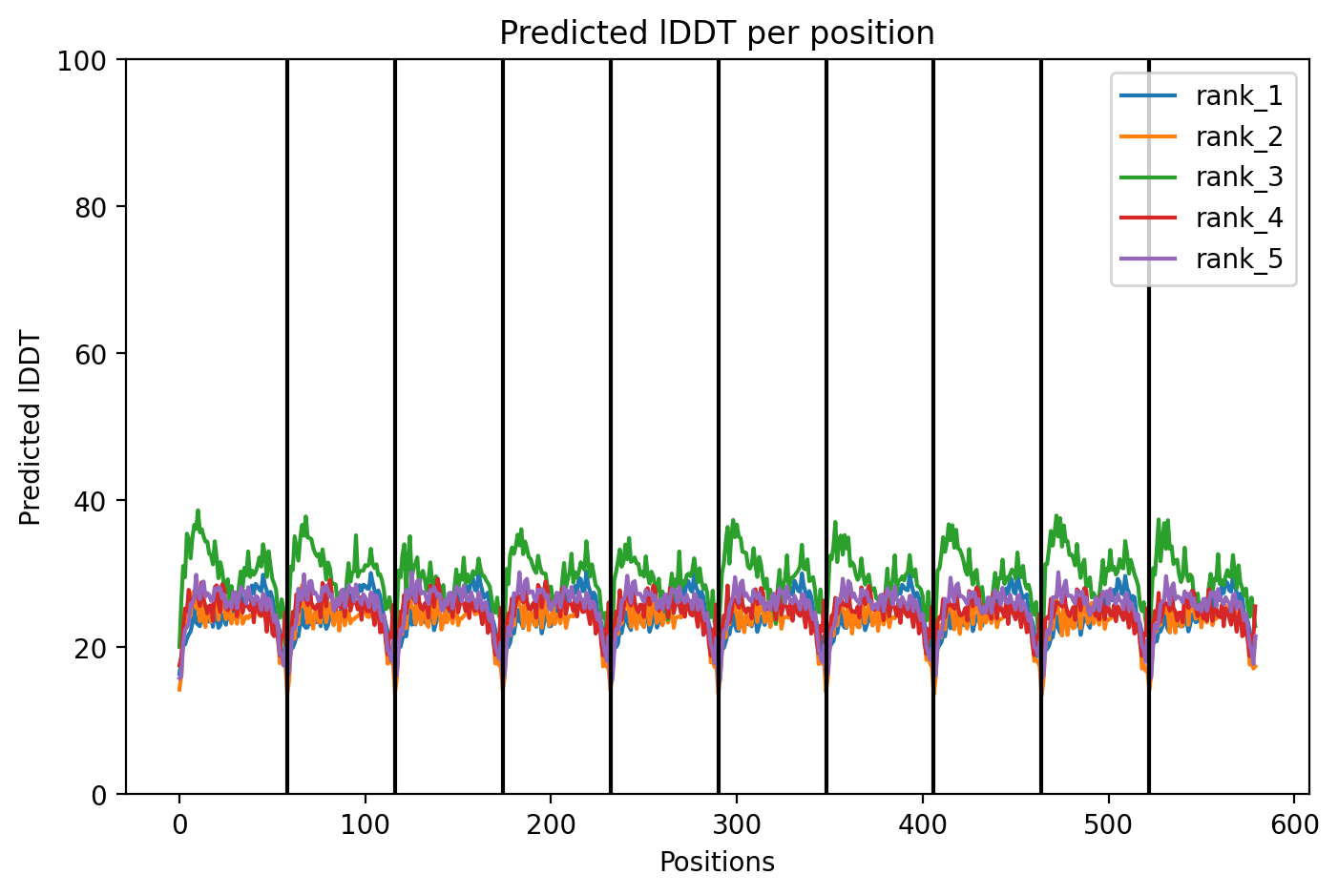
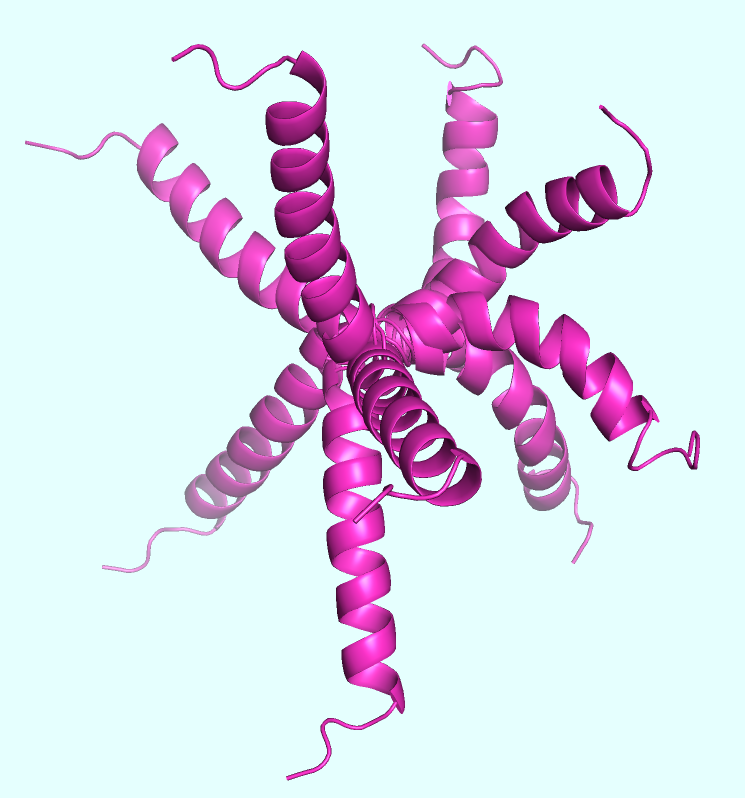
Ниже продемонстрированы структуры (Рис. 7) двух предсказанных моделей: пентамера и декамера. AlphaFold плохо справился с предсказанием амилоидной фибриллы белка α-синуклеина. Быть может, правильнее было бы моделировать не пента- и декамер, а гекса- и додекамер, поскольку в нативном виде именно в 6- и 12-меры собирается выданный белок. Но, думаю, результат от этого сильно не поменялся бы.
Структура фибриллы α-синуклеина кажется мне достаточно нераспространённой, поэтому, видимо, предсказание получилось таким неудачным. И, по-видимому, AlphaFold в принципе с фибриллами справляется несколько менее удачно, чем с глобулярными белками, примеров структур PDB которых - масса (и большая!) в отличие от структур фибриллярных белков, которых немного меньше.

Ссылка на сессию pymol: 4pr_A8.pse.
Задание C: Протеаза и субстрат.
В рамках данного задания необходимо было исследовать, насколько хорошо AlphaFold предсказывает фермент-субстратные взаимодействия на примере протеазы Катепсина K (P43235) и небольшого пептида NSLVAYKA. Катепсин K - цистеиновая протеаза, присутствующая в остеокластах костей. Синтезируется фермент (как и многие протеазы) в виде зимогена, который впоследствии подвергается аутопротеолизу при низком значении pH [6].
В банке PDB была найдена структура профермента катепсина K (1BY8, РСА, 2.60 Å), который в дальнейшем сравнивался с продуктом фантазии AlphaFold.
По результатам структурного выравнивания (Рис. 8) можно сказать, что AlphaFold прекрасно справился с задачей воссоздания структуры белка. Имеется лишь N-концевой неструктурированный участок, который имеет низкую степень уверенности в предсказании.
Далее решено было сделать предсказание уже со зрелым белком, прошедшим этап аутопротеолиза.
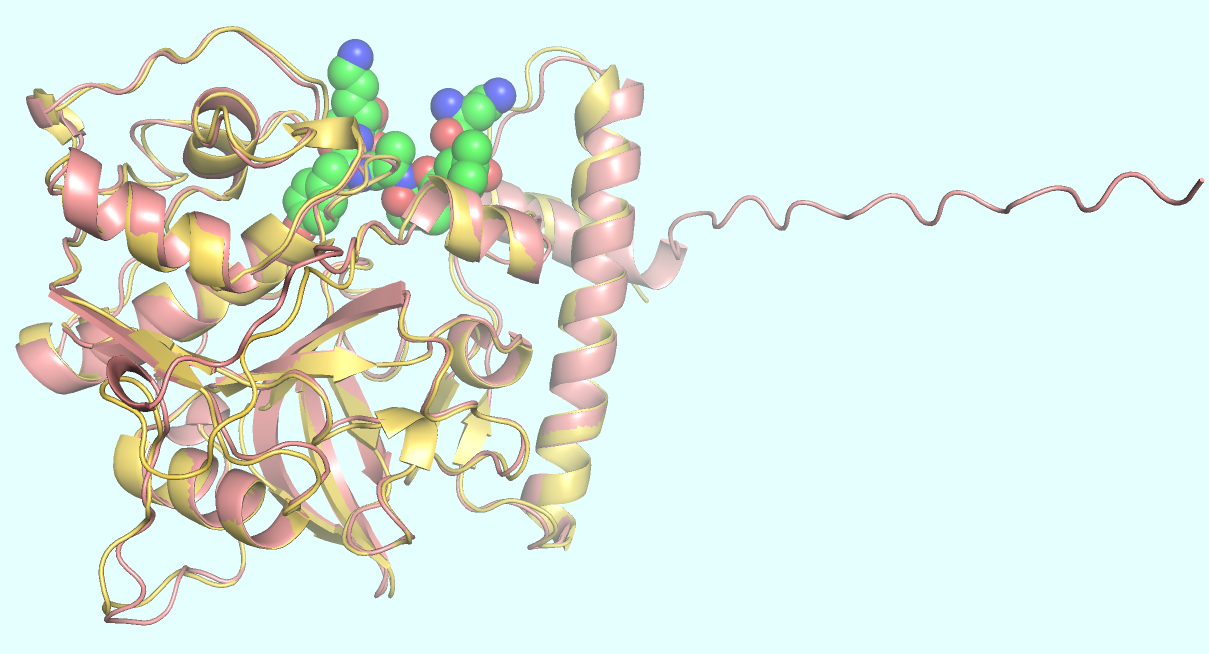
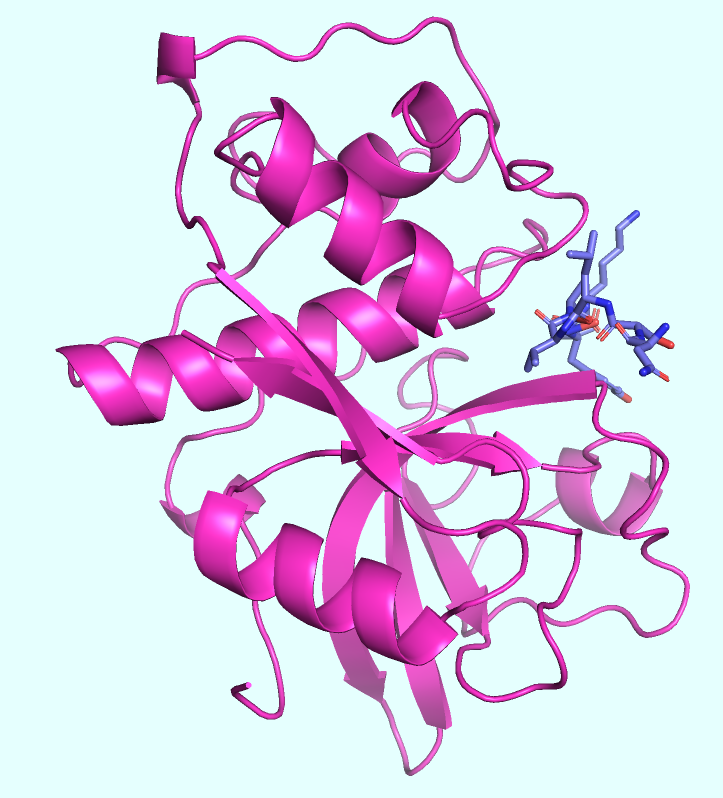
При предсказании "рабочей" версии катепсина K результат оказался отличным (Рис. 9). AlphaFold очень точно предсказал структуру белка (не привожу метрики и структурное выравнивание с экспериментальной версией, но поверьте на слово... =)) и правильно определил место связывания субстрата (небольшого пептида) в активном центре фермента. Это оказалось достаточно неожиданным и приятным сюрпризом.
Ссылка на сессию PyMol: 4pr_C.pse.
Кирилл Кузенков, студент четвёртого курса ФББ