Главная страница
term3 🕒
TASK3&4. Картирование чтений и анализ выравнивания.
TASK6. Аннотация SNP.
term3 🕒
Геномные браузеры
Для выполнения практикума были даны файлы:
chr15.fastq & chr15.fasta
Файлы с резульататми лежат в директории:
/nfs/srv/databases/ngs/kirill
TASK1. Анализ качества чтений.
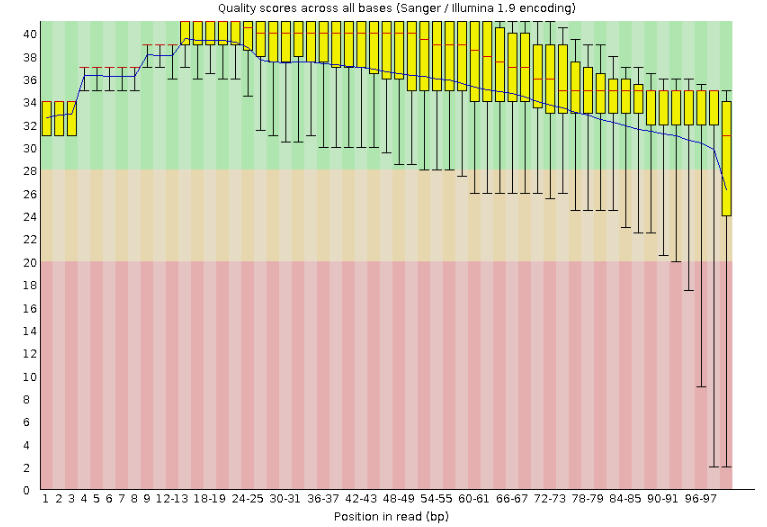
В этом задании использовалась программа FastQC, которая позволяет сделать анализ качества сырых последовательностей,
полученных непосредственно из высокопрозводительного секвенатора.
Команда для вызова программы FastQC: fastqc chr15.fastq
Отчет о программе получен в виде html файла:chr15_fastqc.html
Все фрагменты чтений,кроме последнего, имеют хороший параметр Phred Quality Score.
TASK2. Очистка чтений
В этом задании использовалась программа Trimmomatic, которая производит очистку чтений, а именно
удаление тех фрагментов,длина которых была меньше 50 нукл-в и чьи Q-score меньше 20. Исходя из этих требований
были выбраны trimming steps:
1)MINLEN: Drop the read if it is below a specified length;
2)TRAILING: Cut bases off the end of a read, if below a threshold quality
Команда для вызова программы Trimmomatic:
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr15.fastq trim.fastq TRAILING:20 MINLEN:50
Выходной файл:trim.fastq
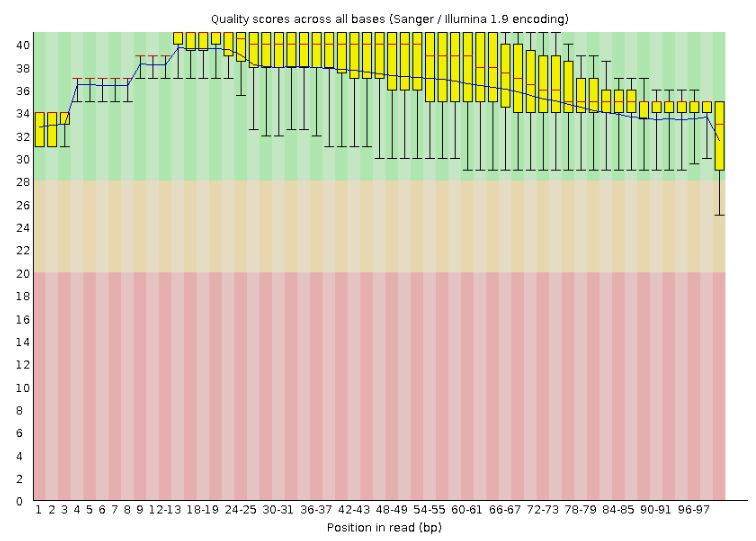
Далее, файл с удаленными фрагментами был проанализирован командой FastQC (fastqc trim.fastq).
HTML-file:c
Число чтений до и после чистки: 5068 и 4946 соответсвенно.
Были удалены те участки, качество нуклеотидов которых ниже 20, и те, длина которых меньше 50 нукл.
|
|
|
TASK3&4. Картирование чтений и анализ выравнивания.
Команда |
Функция |
hisat2-build chr15.fasta chr15 |
Индексирование референсной последовательности,
на выходе получаем файлы chr15.*.ht2, *={1,..,9}
|
hisat2 -x chr15 -U trim.fastq --no-spliced-alignment --no-softclip > align.sam |
Построение выравнивания прочтений и референса в формате .sam Выходной файл align.sam |
samtools view align.sam -b > align.bam |
Перевод выравнивания чтений с референсом в бинарный формат .bam Выходной файл align.bam |
samtools sort align.bam -T align.txt -o sortalign.bam |
Сортировка выравнивания чтений с референсом по координате в референсе начала чтения Выходной файл sortalign.bam |
samtools index sortalign.bam |
Индексация отсортированного sortalign.bam файла Выходной файл sortalign.bam.bai |
samtools idxstats sortalign.bam > info |
Информация о числе чтений, откартированных на геном Выходной файл output file |
(The output is TAB-delimited with each line consisting of reference sequence name, sequence length, # mapped reads and # unmapped reads. It is written to stdout). В файле info представлены числа откартированных(4935) и неоткартированных на хромосому чтений(11), длина последовательности хромосомы(102531392) и ее имя(chr15).TASK5. Поиск SNP и инделей.
Команда |
Функция |
samtools mpileup -f chr15.fasta -g sortalign.bam > snp.bcf |
Создание файла с полиморфизмами в формате .bcf, на выходе получаем файл snp.bcf |
bcftools call -cv snp.bcf -o snp.vcf |
Создание файла со списком отличий между референсом и чтениями в формате .vcf на выходе получаем файл snp.vcf |
№ |
Координата |
Тип полиморфизма |
Буква референса |
Буква чтений |
Качество чтений |
Глубина покрытия |
1 |
89363866 |
замена |
А |
С |
9.52546 |
1 |
2 |
77777632 |
замена |
С |
Т |
225.009 |
49 |
3 |
77721225 |
замена |
А |
G |
9.52546 |
1 |
Следующее задание заключалось в аннотировании полученных SNP при помощи программы ANNOVAR. Для этого был подготовлен файл, который не содержал инделей snp.avinput из snp.vcf c использованием скрипта annotate_variation.pl. Использованная команда: perl /nfs/srv/databases/annovar/convert2annovar.pl.old -format vcf4 snp.vcf > snp.avinput Выходной файл:snp.avinput Далее полученный файл использовался для аннотации SNP по базам данных refgene, dbsnp, 1000 genomes, GWAS и Clinvar c помощью скрипта annotate_variation.pl. Обнаружено 89 полиморфизмов, в числе которых 2 инделя; 87 - замены Качество у найденных полиморфизмов равно 63.00807.(среднее значение) и 11.3429(медиана),что не является высоким показателем. Cреднее покрытие равно 11.12 Медиана покрытия равно 1 Покрытие плохое. refgene Тип аннотации: gene-based annotation. Использованная команда: annotate_variation.pl -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb.old/ Было получено три выходных файла: rs.refgene.variant_function(contains annotation for all variants) rs.refgene.exonic_variant_function(contains the amino acid changes as a result of the exonic variant) rs.refgene.log(contains information about ANNOVAR programm and command working report) SNP может находиться в одном из следующих регионов последовательности: Value Default precedence Explanation Sequence Ontology
exonic 1 variant overlaps a coding exon_variant (SO:0001791)
splicing 1 variant is within 2-bp of a splicing junction splicing_variant (SO:0001568)
ncRNA 2 variant overlaps a transcript without coding annotation in the gene definition non_coding_transcript_variant (SO:0001619)
UTR5 3 variant overlaps a 5' untranslated region 5_prime_UTR_variant (SO:0001623)
UTR3 3 variant overlaps a 3' untranslated region 3_prime_UTR_variant (SO:0001624)
intronic 4 variant overlaps an intron intron_variant (SO:0001627)
upstream 5 variant overlaps 1-kb region upstream of transcription start site upstream_gene_variant (SO:0001631)
downstream 5 variant overlaps 1-kb region downtream of transcription end site (use -neargene to change this) downstream_gene_variant (SO:0001632)
intergenic 6 variant is in intergenic region intergenic_variant (SO:0001628) The first column tells whether the variant hit exons or hit intergenic regions, or hit introns, or hit a non-coding RNA genes. If the variant is exonic/intronic/ncRNA, the second column gives the gene name (if multiple genes are hit, comma will be added between gene names);if not, the second column will give the two neighboring genes and the distance to these neighboring genes. Было получено 89 SNP: exonic=12 intronic=62 UTR=1 upstream=3 ncRNA=10 intergenic=1 Σ=89 Качество у найденных полиморфизмов зависит от процент.числа snp,попавших в категорию exonic.В данном случае таких snp 13%. Мои snp попали в следующие гены:LIPC,LOC101928694,HMG20a,ACAN,AQP9. описание трех полиморфизмов: № Координаты (*) Ген Н. замена Тип замены Качество чтений Глубина покрытия А.к. замена 1 58830707 LIPC C>T het 225.009 63 H88H 2 58834741 LIPC G>T het 201.009 26 V155V 3 89382129 ACAN C>A het 95.0077 11 D102E Dbsnp Тип аннотации: filter-based annotation Команда:annotate_variation.pl -filter -out db.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb.old/ Было получено три выходных файла: db.snp.hg19_snp138_dropped(contains variants that are annotated in dbSNP, and print out their rs identifiers) db.snp.hg19_snp138_filtered(contains SNPs not in dbSNP) db.snp.log(contains information about ANNOVAR programm and command working report) rs имеют 75 snp. 1000 genomes Тип аннотации: filter-based annotation Команда:annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000gen snp.avinput /nfs/srv/databases/annovar/humandb.old/ Было получено три выходных файла: 1000gen.hg19_ALL.sites.2014_10_dropped(contains variants that are annotated in dbSNP, and print out their rs identifiers ) 1000gen.hg19_ALL.sites.2014_10_filtered(contains SNPs not in dbSNP) 1000gen.log(contains information about ANNOVAR programm and command working report) Медиана и среднее значение частоты найденных snp примерно равны (0.478035 и 0.492093 соответственно) Gwas To find variants that were previously reported to be associated with diseases or traits in genome-wide association studies, the gwasCatalog keyword can be used: Тип аннотации: region-based annotation Команда:annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb.old/ Было получено 2 выходных файла: gwas.hg19_gwasCatalog gwas.log(contains information about ANNOVAR programm and command working report) В первом файле представлены 5 snp, которые связанны с определенными заболеваниями или характерной особенностью(в данном случае это cholesterol,Type 2 diabetes,Hematological and biochemical traits,height) Clinvar The ClinVar database archives and aggregates information about relationships among variation and human health. Тип аннотации: filter-based annotation Команда:annotate_variation.pl -filter -out clinvar -build hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb.old/ Было получено три выходных файла: clinvar.hg19_clinvar_20150629_dropped(contains variants that are annotated in dbSNP, and print out their rs identifiers) clinvar.hg19_clinvar_20150629_filtered(contains SNPs not in dbSNP) clinvar.log(contains information about ANNOVAR programm and command working report) Первый файл оказался пустым. Следовательно в данной аннотации не было результатов, связанных с вариациями генома и их отношением к здоровью человека. Таблица с характеристиками snp:EXCEL
© Цыганов Кирилл, 2017