Главная страница
term3 🕒
TASK1. Упражнения по EMBOSS
TASK2. Упражнения по EMBOSS (задача 3)
term3 🕒
EMBOSS & локальный BLAST
TASK1. Упражнения по EMBOSS
| Название упражнения | Команда | Ссылка на результат/исходные данные |
1.Несколько файлов в формате fasta собрать в единый файл. |
seqret @list -out all |
list (1).fasta (2).fasta (3).fasta all |
2.Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы. |
seqretsplit all |
splitted files all |
3.Из файла с аннотированной хромосомой в формате gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле. |
seqret @3 3cds.fasta |
gb file coordinates of coding sequences fasta file |
4.Транслировать (с первого кодона, то есть в первой рамке) кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода, и положить результат в один fasta файл. |
transeq -table 0 -sequence 5.fasta -outseq 4.fasta |
fasta-file with coding sequence translated coding sequences |
5.Вывести открытые рамки длиной не менее заданной, имеющиеся в данной нуклеотидной последовательности. |
getorf 5.fasta -minsize 45 5orf.fasta |
fasta-file open reading frames |
6.Перевести выравнивание из формата fasta в формат msf. |
seqret "(6).fasta" msf::6.msf |
fasta-file msf.file |
7.Выдать в файл число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имена последовательностей и числа). |
infoalign 7.fasta -refseq 2 -onli -name -idcount 71.fasta |
file with alignments result |
8.(featcopy) Перевести аннотации особенностей из файла формата gb или embl в табличный формат gff. |
featcopy sequence.gb sequence.gff |
sequence.gb sequence.gff |
9.(extractfeat) Из данного файла с хромосомой в формате gb или embl получить fasta файл с кодирующими последовательностями. |
extractfeat -type CDS 9.gb 9.fasta |
file.gb file.fasta |
10.Перемешать буквы в данной нуклеотидной последовательности. |
shuffleseq (1).fasta (1)shuf.fasta |
(1).fasta (1)shuf.fasta |
11.Создать три случайных нуклеотидных последовательностей длины 100. |
makenucseq -amount 3 -length 100 -outseq 11 |
file with 3 sequences |
12.Найти частоты кодонов в данных кодирующих последовательностях. |
cusp all 12.fasta |
coding sequences codon's frequency |
13.(tranalign) Выровнять кодирующие последовательности соответственно выравниванию белков — их продуктов |
tranalign -asequence 1pr.fasta -bsequence 2pr.fasta -outseq 13.fasta |
coding sequences aligned prot_sequences aligned nucl_sequences |
14.Удалить символы гэпов из выравнивания (превратив его тем самым снова в набор невыровненных последовательностей). |
degapseq 14.fasta > 14degap |
with gaps without gaps |
15.Перевести символы конца строки из формата Windows в формат Unix. |
noreturn windows.txt unix.txt -system unix |
windows.txt unix.txt |
16.Файл с ридами sra_data.fastq в формате fastq перевести в формат fasta. |
seqret "sra_data.fastq" fasta::16.fasta |
fastq-file fasta-file |
TASK2. Упражнения по EMBOSS (задача 3)
Найти частоты динуклеотидов в геноме бактерии, сравнить их с ожидаемыми и определить динуклеотид, частота которого наиболее отклоняется от наблюдаемой. Подсказка: ожидаемая частота XY = (наблюдаемая частота X) * (наблюдаемая частота Y). сценарий input-file output-file
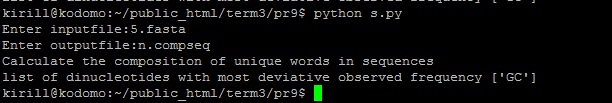
результат сценария
© Цыганов Кирилл, 2017